
皮肤型红斑狼疮的诊治进展
2021-04-15 05:58邓丹琪杨滨宾
诊断学(理论与实践) 2021年1期
邓丹琪,杨滨宾
(昆明医科大学第二附属医院皮肤性病/风湿免疫科,云南 昆明 650101)
红斑狼疮(lupus erythematosus,LE)是一类慢性、反复发作的非器官特异性自身免疫病,其为病谱性疾病,70%~85%的患者有皮肤受累,其疾病谱的一端为皮肤型红斑狼疮(cutaneous lupus erythematosus,CLE),病变主要累及皮肤,另一端为系统性红斑狼疮(systemic lupus erythematosus,SLE),病变可累及多系统和多脏器,几乎全身各系统都可受累[1]。CLE 的临床表现可从局限性的盘状损害到全身泛发性的皮肤病变。目前,CLE 诊断主要依赖于临床表现和实验室检查以及皮肤病理学检查,皮肤红斑狼疮面积和严重程度指数 (cutaneous lupus area and severity index,CLASI) 是衡量疾病活动和皮肤损害(皮损)的有效指标。
CLE 的发病机制涉及多种因素,包括遗传、表观遗传学和环境因素,特别是紫外线的影响。CLE发病的分子机制与细胞因子级联反应的免疫失调及细胞信号通路异常密切相关。目前,CLE 的一线治疗药物主要是抗疟疾(抗疟)药物和糖皮质激素,而免疫抑制剂、沙利度胺和阿维A 可用于顽固性CLE 的治疗。单克隆抗体靶向药物,特别是近年的Ⅰ型干扰素(interferon Ⅰ,IFN-Ⅰ)抑制剂,其临床试验为难治性CLE 患者的治疗带来希望。目前,我国CLE 的诊断率不高,主要与临床医师缺乏规范的诊疗思路有关,加强临床医师对不同类型LE 皮损的认识,有助于早期诊断、正确治疗及改善预后。本文就近年CLE 的发病机制研究新进展及诊治流程进行阐述及总结。
流行病学研究
我国目前缺乏针对CLE 详细的流行病学调查资料。在欧洲和美国,CLE 的发病率为4.2/10 万,略高于SLE 的发病率(3/10 万)。女性的发病率高于男性(5.8/10 万比2.4/10 万),发病年龄在30~69 岁之间。CLE 的患病率为70.4/10 万,女性患病率亦较男性高 (85.1/10 万比56.9/10 万),患病年龄高峰在50~59 岁[2]。慢性皮肤型红斑狼疮(chronic cutaneous lupus erythematosus,CCLE)和亚急性皮肤型红斑狼疮 (subacute cutaneous lupus erythematosus,SCLE)患者因无重要脏器受累,预后大多良好,而急性皮肤型红斑狼疮 (acute cutaneous lupus erythematosus,ACLE) 患者的预后则取决于重要脏器的受累程度。
CLE 的发病机制
CLE 的发病机制涉及多因素,除上述提及的因素,还包括先天性和适应性免疫反应的异常。目前的观点认为,CLE 的致病途径除了树突细胞激活、T细胞失调、细胞因子失衡、B 淋巴细胞缺陷和自身抗体产生外,还有紫外线照射刺激角质形成细胞产生固有免疫相关的细胞因子,并触发细胞死亡,从而激活核酸信号通路。总体而言,目前开展的发病机制研究针对SLE 的相对较多,针对CLE 的则较少些,以下总结近年来的相关发病机制研究。
一、遗传因素
1.TREX1 突变:CLE 常发生于家庭成员和双胞胎之间,提示遗传因素起重要作用。迄今为止,仅鉴定出一种CLE 单基因突变,这是一种罕见的以TREX1 突变为特征的家族性冻疮样红斑狼疮(chilblain lupus erythematosus,CHLE)[3]。TREX1 是一种细胞内3'-5'DNA 外切酶,TREX1 缺乏会导致细胞质DNA 在细胞内过度积累,继而激活IFN-Ⅰ通路,诱导产生更多的CXC 趋化因子配体10(CXC motif chemokine ligand 10,CXCL10),从而引发强烈的自身免疫反应。TREX1 相关的家族性CHLE 患者,从出生起即存在核酸代谢障碍,与散发性发病的CHLE 患者相比,家族性CHLE 患者血清中IFN-Ⅰ和CXCL10 呈高表达,提示IFN-Ⅰ通路呈慢性、持续性的激活状态[4]。
2.其他遗传位点:研究者在不同的CLE 人群中还发现了一些其他的遗传关联、基因突变和基因多态性,如ITGAM 多态性与SLE 皮肤受累密切相关,其中ITGAM 的rs1143679 单核苷酸多态性(single nucleotide polymorphism,SNP) 与ACLE 和盘状红斑狼疮(discoid lupus erythematosus,DLE)相关[5]。ITGAM 编码Mac-1 整合素的CD11b 链,这是一种参与单核细胞、巨噬细胞及中性粒细胞相互作用的表面受体。整合素功能失调可能会阻止细胞的黏附和吞噬,从而导致细胞外基质的清除障碍和免疫系统的激活[2]。
最近一项全基因组研究发现了4 个新的CLE易感位点[6]。该研究共有183 例CLE 患者,其中CCLE 占44.8%,SCLE 占40.4%,肿胀性红斑狼疮(tumid lupus erythematosus,TLE)占14.2%,另有1 288 名健康对照者被纳入研究,研究发现了4 个位于第6 号染色体主要组织相容性复合体(major histocompatibility complex,MHC) 区的SNP 位点(rs2187668、rs9267531、rs4410767 和rs2187668),都位于MHC 区域中涉及抗原呈递、细胞凋亡、DNA处理和IFN 免疫应答等系统功能相关基因附近[6]。
二、环境因素
在遗传易感人群中,不同的环境因素可以激活先天性和适应性免疫应答,并诱导CLE 皮肤病变的发展。紫外线照射是公认的CLE 激发因素,60%~80%的SLE 患者伴有光敏性皮肤病变[7]。一项前瞻性研究发现,紫外线照射可上调CLE 患者皮肤中IFN 相关基因和MHC 相关基因的表达,但在健康个体皮肤中并未观察到此现象[8]。紫外线照射会引发细胞损伤,促进炎症反应,包括细胞死亡、活性氧释放和异常的DNA 修饰[9]。在CLE 小鼠模型中,紫外线照射会导致角质形成细胞死亡,并引发其表面核抗原暴露增加,促进免疫复合物沉积,随后被循环中的自身抗体识别。此外,紫外线照射会刺激肥大细胞释放IFN-α、白细胞介素(interleukin,IL)-1、IL-6、肿瘤坏死因子(tumor necrosis factor,TNF)-α等多种炎症因子,促进炎症,损伤组织[10]。此外,紫外线照射能上调细胞内黏附分子的表达和CXCL10分泌,促进免疫细胞向皮肤归巢[11]。总之,紫外线能通过细胞因子和黏附分子增加、T 淋巴细胞活化、细胞凋亡增加、DNA 甲基化异常等途径诱发和加重CLE 的病理过程。
三、免疫因素
CLE 发病相关的免疫异常主要包括树突细胞异常激活、T 淋巴细胞调节异常、细胞因子失衡、B淋巴细胞缺陷和自身抗体产生。
在CLE 患者中,皮肤损伤机制是抗上皮细胞毒性免疫反应,该反应促进细胞碎片的释放并重新激活先天性免疫应答,从而导致炎症的级联反应。研究表明,角质形成细胞可通过自身产生IFN-Ⅰ和IFN-Ⅲ以及IFN 调节的炎症细胞因子[IFN-α/β、IFN-λ1 (IL-29)、IFN-λ2 (IL-28a)、IFN-λ3(IL-28b)、TNF、IL-12 和IL-6]和趋化因子(CXCL9、CXCL10 和CXCL11)参与炎症和自身免疫损伤[7,12]。
T 淋巴细胞、B 淋巴细胞、树突细胞和巨噬细胞之间的相互作用是经IFN 分泌的大量细胞因子和趋化因子调节完成,其中CXCL10 发挥重要作用。这些促炎介质可通过趋化因子梯度来调节细胞对皮肤血管壁的黏附,并向表皮迁移,继而细胞毒性效应细胞攻击皮损处的角质形成细胞,导致角质形成细胞凋亡和炎症细胞因子的表达、释放,加剧了皮损处的炎症。
CLE 的诊断流程及各分型的临床特点及组织病理学表现
一、诊断
1.诊断流程:CLE 的诊断主要根据各型的临床表现、组织病理学特征以及实验室检查等,具体诊断流程见图1。对临床表现典型的患者,经系统检查排除SLE 后,则可以诊断为CLE;如果患者的临床表现疑似,可以进行病理学检查,同时进行系统检查以排除SLE。当组织学检查发现病理表现符合LE 但不确定,可行直接免疫荧光(direct immunofluorescence,DIF)检测,如在皮损处表皮与真皮交界处见IgG、IgM、IgA 和(或)补体C3呈颗粒状沉积,即为DIF 阳性,此时结合临床表现即可确诊或拟诊CLE,再进一步探查患者有无全身受累。如患者的DIF 检测结果呈非特异性表现或阴性,需进行个体化分析,观察是否存在假阴性结果。在CLE 活动性皮损中,DIF 的阳性率较高;TLE 的DIF 阳性率低;在狼疮性脂膜炎中,DIF 可显示真皮脉管周围的免疫反应物沉淀,但真皮与表皮交界处颗粒状沉积物不会一直存在,导致可能会出现假阴性的结果。若组织学发现与LE 表现不符合,可暂时排除诊断,需定期复查(每6~12 个月复查)血尿粪常规、红细胞沉降率、免疫球蛋白、补体、抗核抗体(antinuclear antibody,ANA)、抗双链 DNA (double-stranded DNA,ds-DNA)抗体及抗ENA 抗体谱。
2.鉴别诊断:ACLE 重点应与玫瑰痤疮、皮肌炎及其他皮肤血管炎等鉴别。SCLE 应与Sweet 综合征、多形性日光疹、银屑病、多形红斑、环状肉芽肿等鉴别。不典型的CCLE 应根据具体的分型与相关疾病鉴别,如DLE 要注意与白癜风、寻常狼疮等鉴别;TLE 要注意与荨麻疹性血管炎等鉴别。
3.实验室检查:LE 患者血循环中具有针对自身组织器官、细胞及其成分的多种抗体。ANA 几乎见于绝大多数SLE 患者,但其特异性低,滴度不一定与疾病活动性相平行。抗dsDNA 抗体是诊断SLE 的标志性抗体之一,多出现在SLE 的活动期。ACLE 与SLE 密切相关,与SLE 相关的特异性抗体在患者中出现的比例高,如ANA (70%~80%)、抗ds-DNA 抗体(30%~40%)。不同类型CCLE 患者的实验室检查如血常规、尿常规多数正常,4%~20%的患者ANA 可以呈低滴度阳性;1%~3%的患者有抗SSA(Ro)抗体阳性;<5%的患者出现抗dsDNA 抗体。Biazar 等[13]的一项研究发现,ACLE、SCLE、DLE 患者血清抗SSA 抗体阳性率分别为47.4%、72.1%、22%;血清抗SSB(La)抗体阳性率分别为27.5%、36.2%和7%。一项针对白种人SLE 患者的分析表明,抗Sm 抗体与盘状皮疹及光敏性之间存在关联;抗SSA 抗体与面颊部皮疹、口腔溃疡及类风湿因子间存在关联[14]。另一项主要针对中国SLE 患者的研究表明[15],抗SSA 抗体、抗SSB 抗体与光敏性、盘状损害密切相关;抗Sm 抗体、抗核蛋白抗体、抗磷脂抗体与面颊部皮疹、黏膜损害、浆膜炎及关节炎相关;抗ds-DNA 抗体与肾脏受累密切相关。这些不一致的发现可能是由于种族背景的差异,需要更多的研究来阐明自身抗体与疾病临床表现之间的潜在关联。
4.疾病活动评估:CLASI 是一种专门用于评估CLE 疾病活动和损害的指标[16]。CLASI 也是评价CLE 治疗效果的可靠指标,其活动评分较基线下降20%代表治疗有效,20%是区分患者对治疗有无反应的判断标准[17]。
5.组织学病理:ACLE、SCLE 和DLE 三者共同的病理学改变包括皮损处不同程度的过度角化和基底细胞液化、变性及真皮水肿,真皮与表皮交界处的单核细胞浸润可延伸至真皮。但在SCLE 中,基底细胞变性可以是灶性的,但液化更显著;真皮见浅层单核细胞浸润,真皮与表皮交界不清楚,少数还可见到表皮坏死;细胞浸润通常局限于血管周围和真皮上三分之一的附件结构,表皮可有轻度萎缩。真皮、表皮交界处的液化变性有时会产生囊泡样改变,这在环形SCLE 活动性皮损的边界处特别明显。与ACLE、SCLE 相比,DLE的角化过度更严重,附属器的单核细胞浸润更为明显,毛囊口有角质栓形成,真皮中还可见噬黑素细胞[10]。
二、分型及临床表现
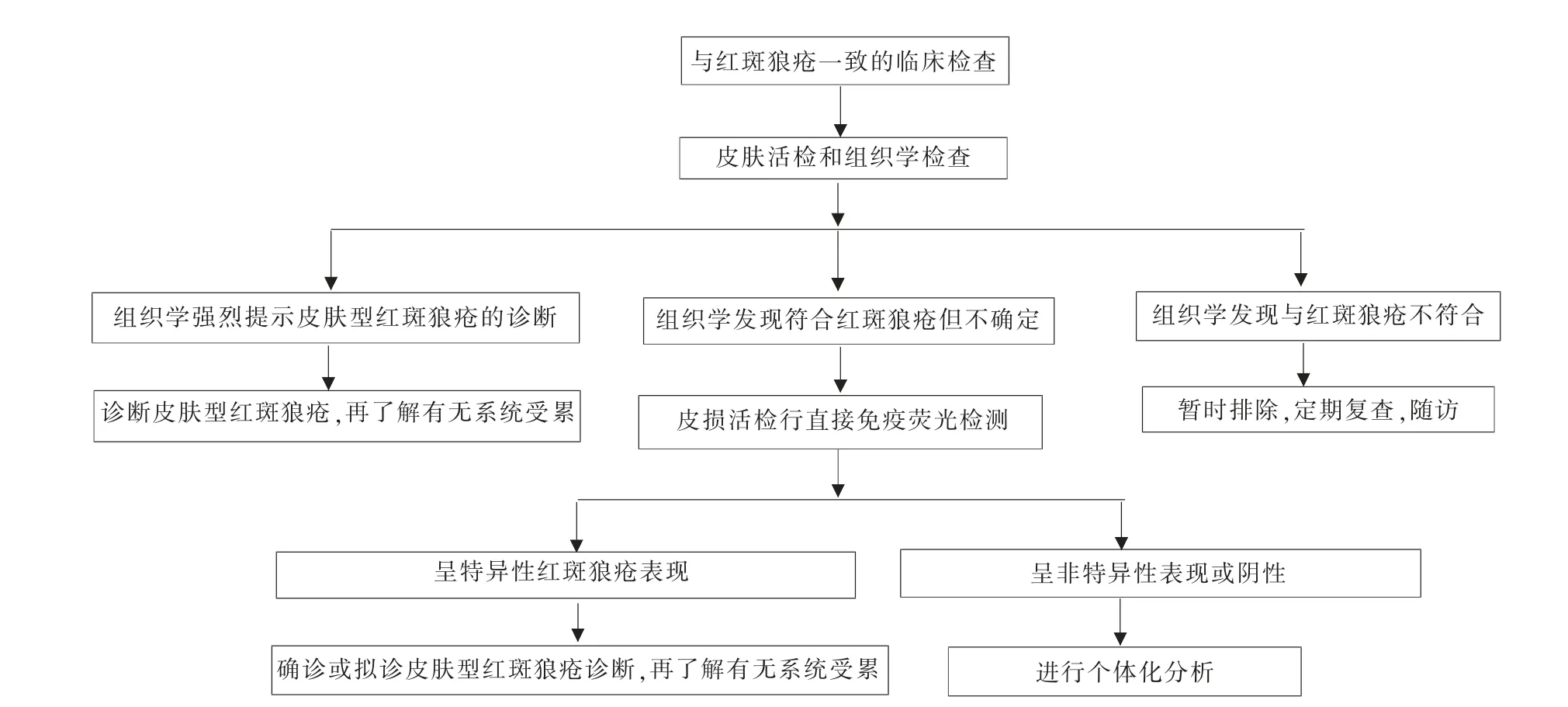
图1 CLE 诊断流程
CLE 根据皮肤病变表现可分为以下类型。①ACLE,包括局限性和泛发性;②SCLE,包括环形红斑型和丘疹鳞屑型;③CCLE,包括局限性和播散性DLE、疣状红斑狼疮(verrucous lupus erythematosus,VLE)、TLE、深在性红斑狼疮 (lupus erythematosus profundus,LEP)、CHLE、Blaschko 线状红斑狼疮。其他罕见的CLE 包括大疱性急性红斑狼疮(以表皮下大疱为特征)、Rowell 综合征 (多形红斑样靶型损害)、新生儿红斑狼疮(neonatal lupus erythematosus,NLE)和皮肤黏膜性红斑狼疮[伴有口腔溃疡、斑块和(或)盘状病变][17]。
1.ACLE:局限性皮疹表现为位于面颊部、鼻背部的融合性水肿性红斑(蝶形红斑)(见图2A);泛发性ACLE 皮疹表现为全身对称分布的融合性斑疹、丘疹,可发生于身体的任何部位,主要累及日晒部位。ACLE 与SLE 密切相关,大多数患者可发展为全身性疾病。
2.SCLE:皮疹表现主要包括丘疹鳞屑型和环形红斑型皮损(见图2B)。皮肤病变主要发生在颈部、肩部、手臂和(或)腿部日晒的皮肤区域。与紫外线相关的自身抗体中,SCLE 患者抗SSA 抗体阳性率为70%~80%,抗SSB 抗体阳性率为30%~40%。20%~30%的SCLE 患者符合诊断SLE,患者中肾脏损害和关节炎最常见[18]。
3.CCLE:患者主要以皮肤损害为主,实验室检查大多正常,临床病程长(数月至数年)且进展缓慢,大部分预后较好。根据皮损表现常分为以下6型,其系统治疗相似,局部治疗因不同分型有所不同,详见后文治疗部分。
DLE 皮损特征为边界清楚的盘状红斑、斑块,表面黏附鳞屑,剥离鳞屑可见背面扩张的毛囊口形成毛囊角质栓,外周色素沉着,中央色素减退、轻度萎缩(见图2C)。DLE 可以是局部性的,也可以是播散性的。高达28%的DLE 患者可发展为SLE,70%的DLE 患者在发病后5 年内发展为SLE。若DLE患者出现弥漫性非瘢痕性脱发、甲周毛细血管扩张、雷诺现象和皮肤血管炎等非特异性LE 皮损和全身性淋巴结肿大,则其发展为SLE 的可能性较大[15]。
VLE 皮损表现为广泛的角化过度,肥厚甚至呈疣状,皮损表面覆盖有多层角质性白黄色鳞屑或厚痂;好发于肢体伸侧和摩擦部位,如上肢伸侧、手和面部(见图2D)。此型多见于DLE 长期未愈的患者。
LEP 是一种少见的亚型,皮损累及皮下脂肪组织,严重时会导致硬结性斑块,并引起局部损毁和凹陷(见图2E),部分可发展为SLE 或发生于SLE 患者。
CHLE 皮损由红色或暗紫红色丘疹和斑块组成,常分布于肢端、耳朵和鼻子(见图2F)。寒冷(特别是潮湿阴冷的气候)可引发或加重症状。
TLE 皮损为多环状隆起性红斑或风团样斑块,表面光滑,无鳞屑和毛囊角质栓(见图2G)。皮损好发于面部或肢体,光敏感明显。
Blaschko 线状红斑狼疮是一种少见的CCLE,皮损多为沿Blaschko 线分布的红斑、皮下结节,好发于头面部(见图2H),少有光敏感现象[15]。
CLE 治疗进展
充分了解不同的CLE 的分型、临床特点、组织病理能更好地指导治疗。迄今为止,还没有一种药物被批准专门用于CLE 的治疗。根据中华医学会皮肤性病学分会红斑狼疮研究中心制定的 《皮肤型红斑狼疮诊疗指南(2019 年版)》,目前CLE 主要采用阶梯治疗,指南侧重于使用局部药物、抗疟药物、糖皮质激素和经典的免疫抑制药物治疗[17]。
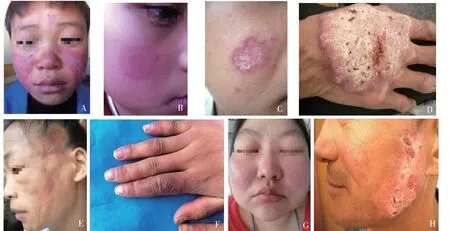
图2 CLE 的分型及临床表现
一、局部治疗
1.防晒:由于紫外线是CLE 皮损重要的诱因之一,因此进行有效防晒至关重要,尤其是色素减少的皮损或者慢性盘状损害,其发生皮肤癌的风险更高[18]。防晒可预防CLE 患者皮肤病变的发生。研究提示,有效防晒可以下调IFN-Ⅰ、IFN-Ⅲ及相关细胞因子 (如TNF 和IL-6) 和趋化因子 (特别是CXCL10)在皮肤中的表达,从而缓解患者的全身炎症[19]。
2.糖皮质激素的局部使用:糖皮质激素具有抗炎作用,是治疗CLE 皮损的一线药物。外用糖皮质激素的主要适应证是局限性DLE,尤其是活动性盘状皮损和肿胀性皮损。皮损内注射曲安西龙非常有效,常用质量浓度为4~5 mg/mL[20]。当皮损顽固时,可每个月重复注射。对全身性DLE 病变和其他CLE 亚型的患者,建议采用系统治疗联合局部激素外用治疗。
3.钙调磷酸酶抑制剂的局部使用:他克莫司软膏和吡美莫司乳膏可用于治疗CLE。钙调磷酸酶抑制剂对SCLE、ACLE 有一定疗效,对DLE 疗效略差。
4.维A 酸类制剂:如他扎罗汀凝胶和维A 酸乳膏等,可用于角化明显的DLE 和VLE。
二、系统治疗
根据2017 年欧洲皮肤型红斑狼疮诊疗指南和我国 《皮肤型红斑狼疮诊疗指南 (2019 年版)》,泛发性及严重CLE 的一线治疗药物是抗疟药物和糖皮质激素。
1.抗疟药物:抗疟药物是系统治疗的一线用药,对DLE、TLE 和SCLE 的有效率可达80%以上,其作用机制仍在研究中。目前研究主要认为抗疟药物能抑制免疫激活,从而抑制外周血单个核细胞产生IFN-Ⅰ[21]。治疗首选硫酸羟氯喹,使用剂量为每次200 mg,每日1 次或2 次。对于硫酸羟氯喹反应差的患者,可以选用氯喹(125~250 mg/d)[22],但需注意的是,服用硫酸羟氯喹或氯喹的患者,应定期进行眼部检查。
2.糖皮质激素:顽固的DLE、ACLE 以及部分SCLE 需要系统使用糖皮质激素治疗,对于成人患者,推荐糖皮质激素与硫酸羟氯喹联合使用。一般选用中小剂量,如泼尼松0.5 mg/(kg·d),病情控制后缓慢递减并尽早停用。对于无系统受累的CLE患者,不推荐糖皮质激素长期维持治疗。SLE 则参照SLE 诊疗指南治疗。
3.免疫抑制剂和免疫调节剂:对于顽固、慢性反复发作的CLE,可联合其他免疫抑制和免疫调节药物。推荐氨甲蝶呤用于难治性CLE,主要用于SCLE,使用剂量为每周7.5~20.0 mg,与硫酸羟氯喹联合使用,使用过程中应注意观察疗效及不良反应,并及时调整用药[23]。对于不伴有系统受累的CLE 患者,不推荐使用硫唑嘌呤、环孢素A 及环磷酰胺[20]。
4.其他药物:对抗疟药治疗反应较差的患者,可选择口服阿维A、沙利度胺、金制剂、氯法齐明(氯苯吩嗪)、柳氮磺吡啶等药物。某些CLE 患者(尤其是VLE)对其他治疗无反应时,推荐使用维A酸类,如阿维A,或异维A 酸[24]。沙利度胺可用于治疗复发或难治性CLE,推荐将其与硫酸羟氯喹联合使用,成人初始剂量一般推荐为100 mg/d,分2 次口服。氨苯砜可用于治疗顽固性CLE 和大疱性急性红斑狼疮,也用于常规治疗效果不理想的DLE和SCLE,推荐与硫酸羟氯喹联合使用,建议从低剂量开始(50 mg/d),最大剂量不超过1.5 mg/(kg·d),治疗前建议进行葡萄糖-6-磷酸脱氢酶活性以及HLAB*13:01 基因检测[25]。由于防晒(减少维生素D的产生)和应用糖皮质激素(增加骨质疏松风险)的不良影响,2017 年欧洲皮肤病学和性病学学会指南建议所有CLE 患者应补充维生素D[20]。
三、CLE 的靶向治疗
自2011 年美国食品药品监督管理局(Food and Drug Administration,FDA) 批准贝利尤单抗(belimumab)可用于治疗SLE,已有10 年的时间[24],目前靶向治疗主要针对SLE,还没有一种药物专门针对CLE。因此,加强对CLE 分子机制的研究能更好地帮助医师探索有关CLE 的靶向治疗策略。目前,靶向治疗CLE 的研究主要针对免疫细胞 (如B 淋巴细胞、T 淋巴细胞和树突细胞)、免疫反应途径[如模式识别受体(pattern-recognition receptor,PRR)信号通路、Janus 激酶(Janus kinase,JAK)信号通路、信号转导及转录激活蛋白(signal transducer and activator of transcription,STAT) 信号通路和核因子κB信号通路]以及细胞因子和趋化因子(如IFN-Ⅰ、CXCL10、IL-6 和IL-12)等多种免疫异常[26]。
1.贝利尤单抗:贝利尤单抗是一种抗B 淋巴细胞激活因子的单克隆抗体,于2011 年被FDA 批准用于治疗SLE[24]。B 淋巴细胞激活因子是角质形成细胞经PRR 刺激后产生的调节IFN 的细胞因子之一,其可能在皮肤先天性免疫系统和适应性免疫系统之间的反馈回路中发挥重要作用。为评估贝利尤单抗在CLE 中的疗效,目前正在进行Ⅲ期临床试验[27]。
2.利妥昔单抗(rituximab):这是一种抗CD20阳性B 淋巴细胞抗体。尽管在最初的研究中显示,利妥昔单抗治疗对一部分活动性CLE 有效,但更多的研究未能支持该药物对大多数类型CLE 的疗效[28]。
3.其他靶向药物:其他靶向药物包括BIIB059及VIB7734 等。浆细胞样树突细胞(plasmacytoid dendritic cell,pDC)是CLE 固有免疫系统中最重要的免疫细胞。在组织中,pDC 可以通过特异性受体CD303(也称为BDCA-2)的表达来识别抗原,并进一步加工处理和呈递抗原。BIIB059 可与CD303 结合,抑制IFN-Ⅰ及其他炎症因子的产生。BIIB059 目前正在进行用于CLE 治疗的临床试验,初步结果表明,BIIB059 治疗使CLE 的CLASI 活动评分下降[29]。VIB7734 是一种针对白细胞免疫球蛋白样受体亚家族成员4 的单克隆抗体,其仅针对pDC,一项Ⅰ期临床试验正在研究VIB7734 治疗CLE 和相关自身免疫病的疗效[30]。
JAK1 和JAK2 的抑制剂鲁索替尼(ruxolitinib)能在体外抑制CLE 特征性趋化因子 (如CXCL9、CXCL10 和CXCL11)在角质形成细胞中的表达,对CHLE 患者有一定的疗效[31]。目前,JAK1 抑制剂(filgotinib,GLPG0634)正在进行Ⅱ期临床试验。
小结及展望
CLE 患者的皮损经治疗多能消退,部分CCLE可遗留萎缩性瘢痕和色素沉着或脱失,新皮损的出现或原有皮损加重往往提示病情活动。CCLE 和SCLE 患者因无重要脏器受累,预后大多良好,而ACLE 患者的预后取决于重要脏器的受累程度。CLE 的治疗仍然是一个挑战,不同亚型的CLE,甚至同一种亚型CLE 患者对相同治疗的反应也可不同。目前,学者发现了可能导致特定疾病表现的遗传和表观遗传学改变,动物实验亦进一步证实了皮肤炎症与SLE 疾病活动之间的联系。IFN-Ⅰ抑制剂的临床试验将为难治性CLE 的治疗带来希望。深入研究CLE 的分子机制,结合先天性免疫和适应性免疫反应途径的新进展,将进一步加深临床对CLE 发病机制的理解,并为CLE 新的治疗策略提供方向。
猜你喜欢
中国中西医结合皮肤性病学杂志(2022年4期)2022-09-18
中国临床新医学(2022年8期)2022-09-08
北方牧业(2021年18期)2021-10-27
婚育与健康(2021年14期)2021-10-18
皮肤病与性病(2021年3期)2021-07-30
中华养生保健(2021年18期)2021-02-13
作文评点报·作文素材初中版(2019年37期)2019-11-16
祝您健康(2018年12期)2018-11-27
家庭医药(2018年2期)2018-02-09
分析化学(2018年12期)2018-01-22
