
气相色谱法测定菜籽油中溶剂残留的 不确定度评定
2021-04-14 03:03张居舟
现代食品 2021年3期
◎ 杨 梅,王 欣,张居舟
(安徽省食品药品检验研究院食品检验所,安徽 合肥 230051)
菜籽是我国的主要油料之一,其特有的香味和滋味受到人们的喜爱,尤其是湖南、四川等地区的居民。菜籽油已成为我国城乡居民膳食用油的主要品种之一[1-2]。
食用菜籽油一般有两种生产工艺,压榨法和浸出法。压榨法是指用机械压榨的方式从油料中榨取出油,浸出法则是采用萃取原理,用食品级6 号溶剂作为萃取剂,从油料中抽提出油脂[3]。虽然浸出法在生产过程中加入了脱除萃取剂流程,但仍可能有部分溶剂残留,带来了一定的风险。食用植物油中残留的有毒物质主要是苯类化合物,有关研究表明,芳香烃化合物会引起淋巴细胞亚群数量和功能上的失调,导致免疫异常,呼吸中枢麻痹,长期食用含有溶剂的油脂损害人体健康[4]。同时溶剂残留量作为一个重要的质量指标出现在《菜籽油》(GB/T 1536—2004)国家标准中,可见国家对食用油溶剂残留的关注。
测量不确定度是表征合理地赋予被测量之值的分散性,与测量结果相联系的参数。其数值的大小反映了测量方法的可靠性,不确定度越小,说明方法越可靠,越接近真实值[5]。本文依据《测量不确定度评定与表示》[6]和《化学分析测量不确定度评定》[7],对菜籽油中溶剂残留量的不确定度进行评定,为气相色谱法测定菜籽油中的溶剂残留提供科学、完整的数据参考。
1 材料和方法
1.1 试剂和设备
6 号溶剂(10 mg·mL-1),由国家粮食局科学研究院提供。
GC2010PLUS 气相色谱仪(日本岛津公司)、移液器(美国GILSON 公司)、XS4002S 型电子天平(瑞士梅特勒公司)。
其余试剂、材料和设备参见《食品安全国家标准 食品中溶剂残留量的测定》(GB 5009.262—2016)的要求。
1.2 样品前处理方法
按照GB 5009.262—2016 的分析方法进行:称量5.0 g(精确到0.01 g)基体植物油6 份于20 mL 顶空进样瓶中。向每份基体植物油中迅速加入5 μL 正庚烷标准工作液作为内标(即内标含量68 mg·kg-1),用手轻微摇匀。
1.3 气相色谱条件
1.3.1 顶空进样条件
平衡时间:30 min;平衡温度:60 ℃;振荡器转速:250 r·min-1;进样体积:500 μL。
1.3.2 气相色谱条件
色谱柱:HP-5(30.0 m×0.25 mm×0.25 µm);柱温度程序:50 ℃保持3 min,以1 ℃·min-1升至55 ℃保持3 min,30 ℃·min-1升至220 ℃保持3 min。进样口温度:250 ℃;检测器FID 温度:300 ℃;进样模式:分流模式;分流比:100 ∶1;载气氮气流速:1 mL·min-1;氢气流量:25 mL·min-1;空气流量:300 mL·min-1。
1.4 数学模型的建立
按照GB 5009.262—2016的测定方法及结果计算,待测物的含量按式(1)计算。

式(1)中:X为试样中待测组分的含量,单位为mg·kg-1;ρ为标准曲线得到的试样中溶剂残留的含量,单位为mg·kg-1。
考虑到实验过程中各种随机因素对测量结果的影响,在测量模型中引入重复性系数frep,建立油样品中溶剂残留量的不确定度测量模型如式(2)。

2 结果与分析
2.1 不确定度的来源分析
根据本实验的测定过程,对菜籽油中溶剂残留的测定结果有影响的各种不确定度的分量进行分析,具体引入的不确定度来源因果如图1 所示。
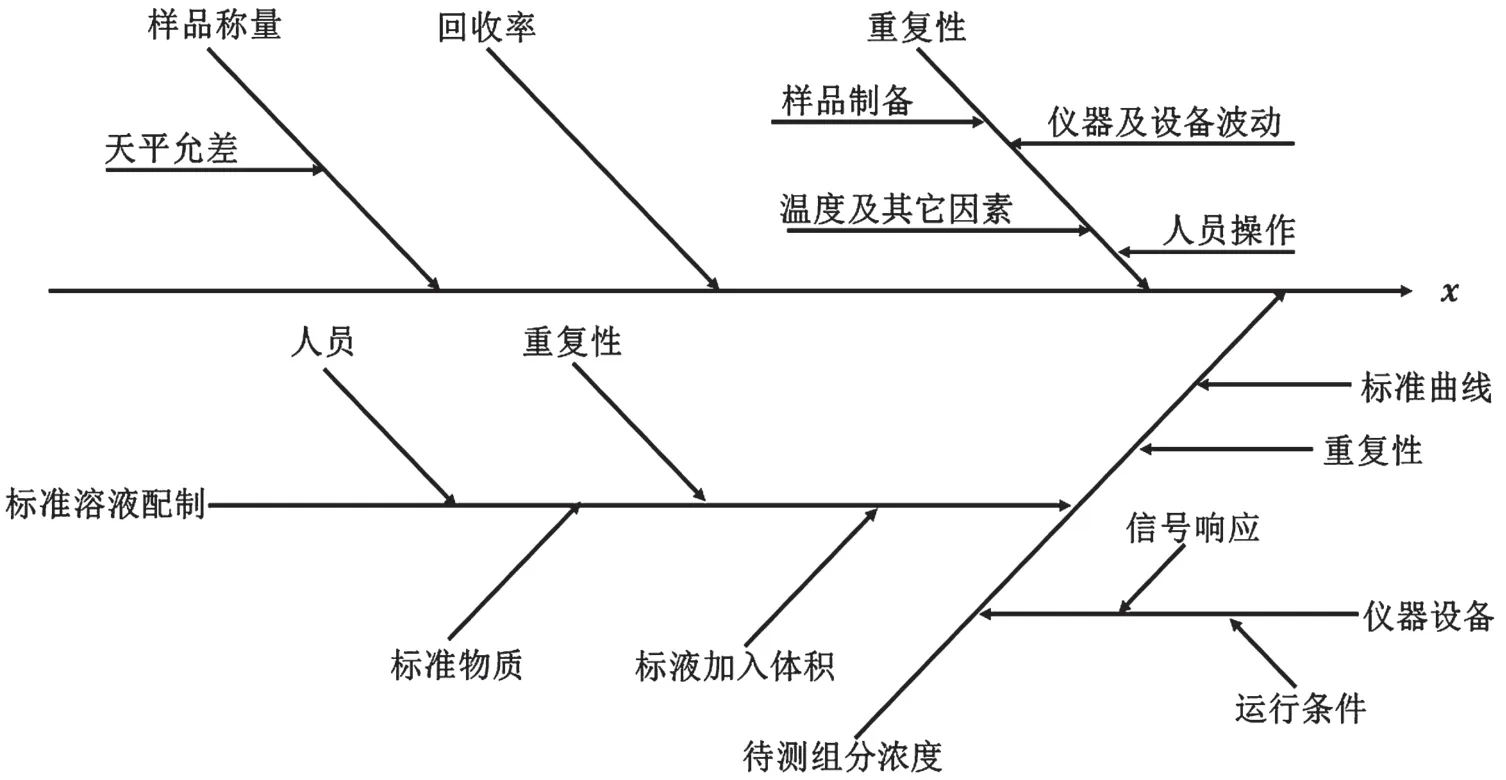
图1 溶剂残留量不确定度来源图
由图1 可知,影响测定结果不确定度urel(X)的主要因素有:称量样品引入的不确定度urel(m)、待测物浓度引入的不确定度urel(C)、整个实验重复性的不确定度urel(frep)以及回收率产生的不确定度urel(R)。
2.2 测量不确定度评定
2.2.1 称量样品引入的不确定度urel(m)
天平引入的不确定度属B 类不确定度,主要由称量时天平的最大允许误差构成。称取5.0 g(精确到0.01 g)菜籽油样品,天平校准证书说明最大允许误差为±0.01 g,按均匀分布考虑,其不确定度u(m)=0.01 g/=0.005 8 g,相对标准不确定度为:urel(m)=u(m)/m=0.005 8 g/5.0 g=0.001 2。
2.2.2 校准过程引入的不确定度urel(C)
校准过程产生的不确定度具体包括标准物质引入、标准系列溶液配制和由标准曲线拟合所产生的不确定度3 个部分。
(1)标准物质引入的不确定度urel(C1)。6 号溶剂标准储备液购于国家粮食局科学研究院,由标准证书可知,浓度为10.0 mg·mL-1(n=10),标准偏差为0.5 mg·mL-1。则6 号溶剂标准溶液标准不确定度为u(C1)=0.5/=0.158 1 mg·mL-1;相 对 不 确 定 度urel(C1)=0.015 8。
(2)标准系列溶液配制过程引入的不确定度urel(C2)。用10 μL、50 μL、100 μL 移液器迅速加入0 μL、5 μL、10 μL、25 μL、50 μL 和100 μL 的6 号溶剂标准品于5 g 空白植物油基质溶液中,加盖密封。得到浓度分别为0 mg·kg-1、10 mg·kg-1、20 mg·kg-1、50 mg·kg-1、100 mg·kg-1和200 mg·kg-1的基体植物油标准溶液。移液器的误差参考JJG 646—2006[8]的要求,按A 类评定,如表1 所示。
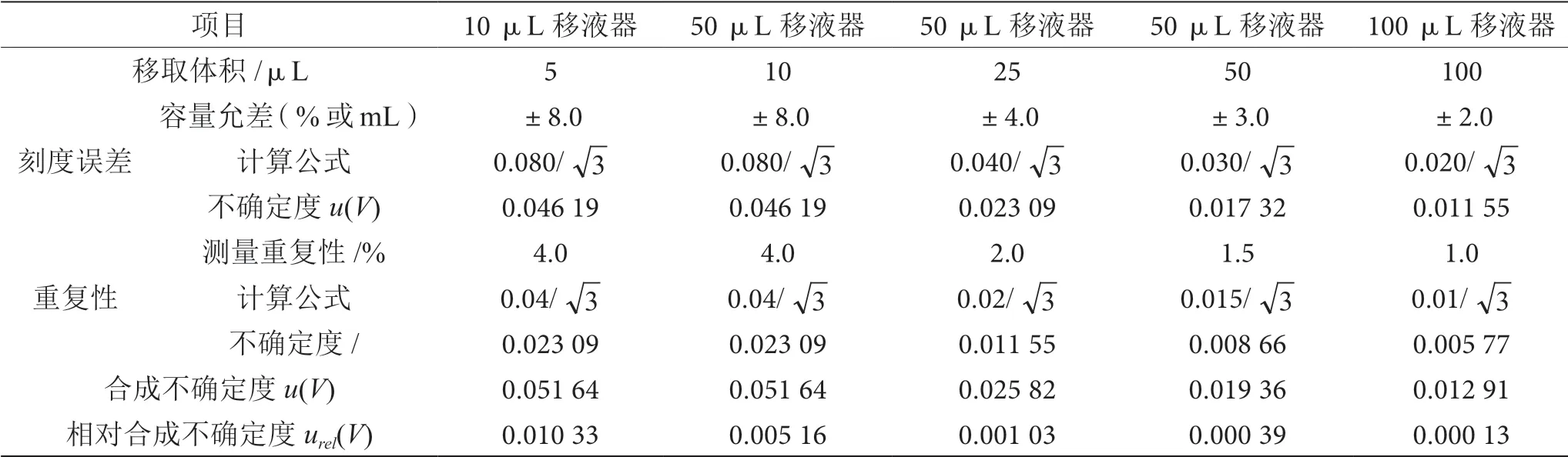
表1 标准系列溶液配制过程引入的不确定度表
则标准系列溶液配制过程引入的相对标准不确定度为
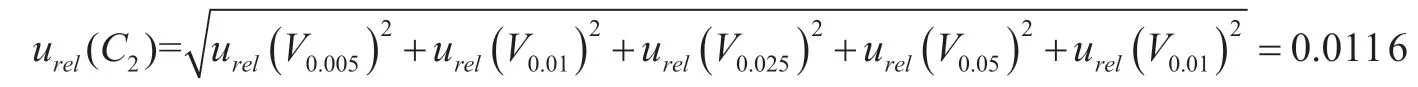
(3)标准曲线拟合产生的不确定度urel(C3)。配制6种标准溶液浓度,分别重复进样3次,以峰面积(Ai)为纵坐标,标准溶液浓度(Ci)为横坐标,采用最小二乘法拟合而成的线性回归方程为Ai=aCi+b,测定数据如下表2 所示。
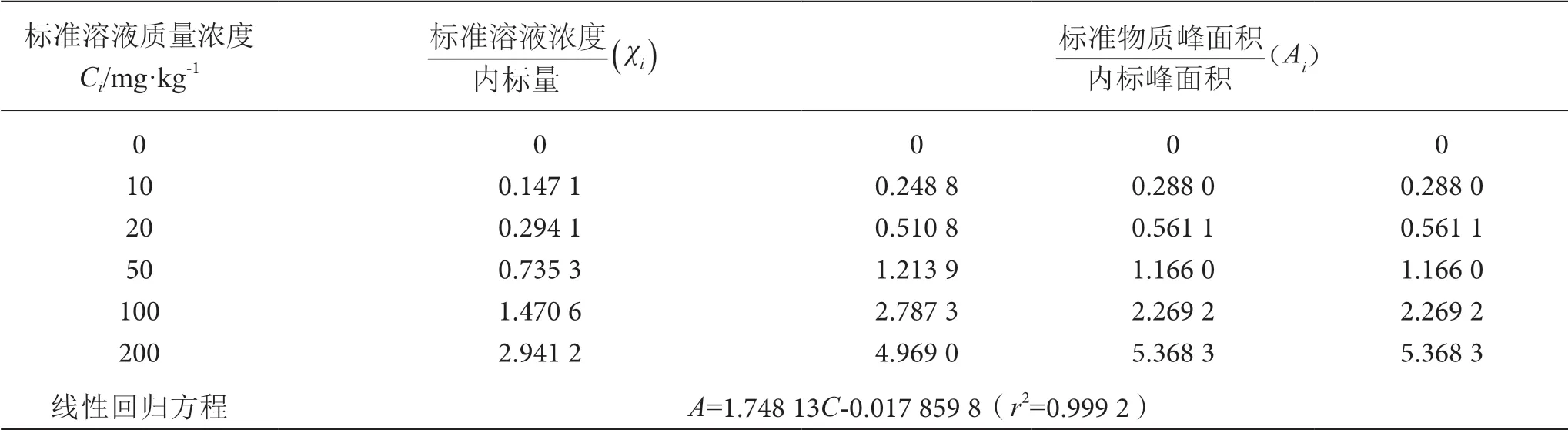
表2 标准曲线的测量结果表
取菜籽油样品进行2 次重复测定,结果分别为8.922 mg·kg-1、8.533 mg·kg-1,平 均 值8.728 mg·kg-1。C0=8.728/68=0.128 4。则由标准曲线拟合产生的不确定度按(3)式计算:
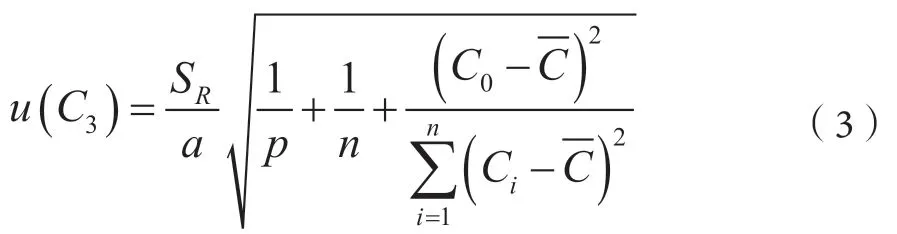
其中

其中:SR-标准溶液待测物与内标峰面积比残差的标准差;Ai-标准溶液各点的待测物的峰面积和内标峰面积的比值;Ci-标准溶液各点浓度与内标浓度的比值;a-拟合直线的斜率;b-拟合直线的截距;n-标准溶液测定次数,本实验室中n=6×3=18 次;p-菜籽油样品测定次数,本实验中p=2;C0-由线性方程求得的样品溶液中待测物与内标浓度比值;-标准溶液各点浓度与内标浓度的比值的平均质量浓度。
则相对标准不确定度urel(C3)=u(C3)/C0,结果见表3。

表3 曲线拟合引入的不确定度相关量表
2.2.3 重复性引入的不确定度urel(frep)
称取菜籽油样品,平行测定6 次,数据见表4。重复测定的含量平均值=8.656 mg·kg-1,标准偏差S()=0.260 03,不确定度则由重复性引起的相对不确定度
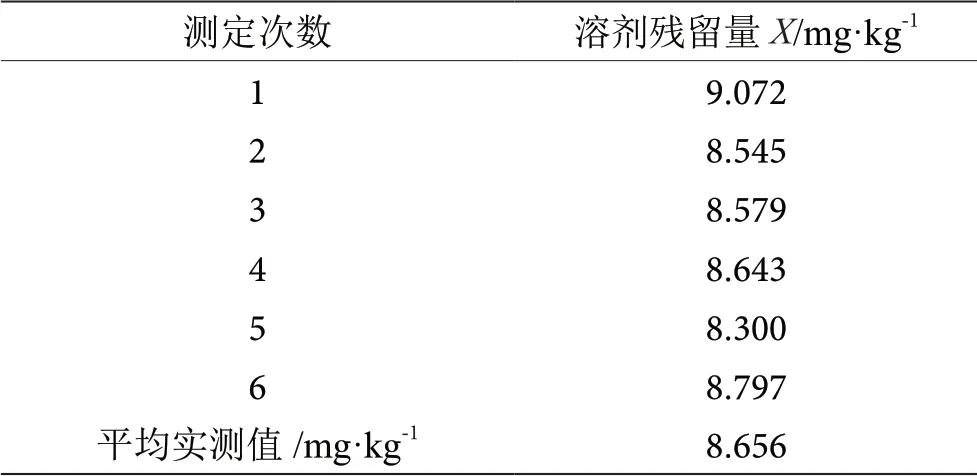
表4 菜籽油中溶剂残留测定结果表
2.2.4 回收率引入的不确定度urel(R)
回收率引入的不确定度按A 类评定,选取空白菜籽油样品,添加浓度水平10.0 mg·kg-1的6 号溶剂标准工作液,平行测定6 次,结果见表5。

表5 菜籽油中添加6 号溶剂的标准回收率结果表
平均回收率=98.6,标准偏差S(R)=3.722 1,
2.3 合成不确定度和扩展不确定度
溶剂残留的相对不确定度分量见表6。不考虑各不确定度的相关性,菜籽油中溶剂残留的合成不确定度为:
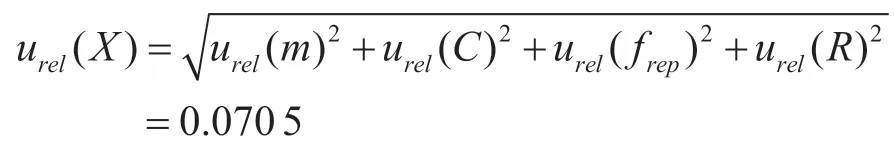
依据《化学分析中不确定度的评估指南》(CNAS-GL006-2018), 在95% 置 信 区 间, 对 于大多数测量选择包含因子k=2,则扩展不确定度
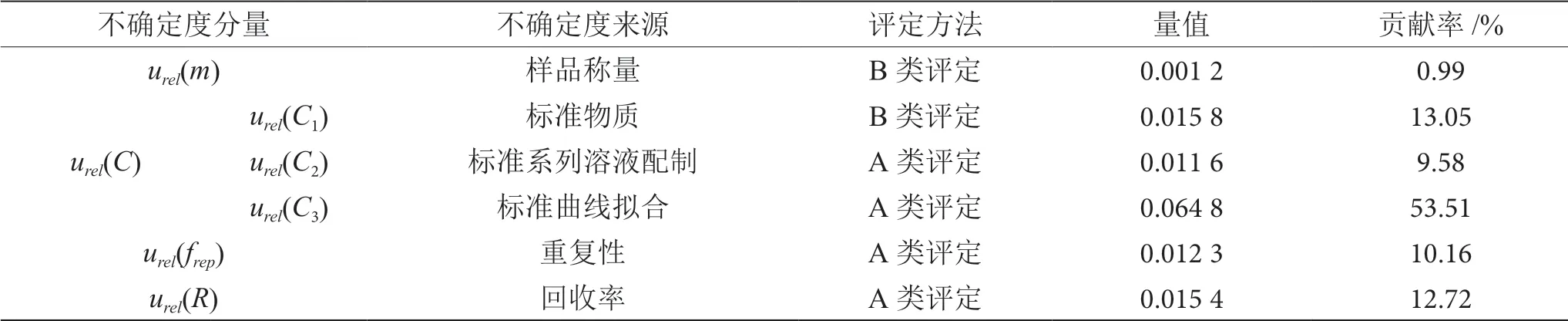
表6 溶剂残留相对不确定度分量表
2.4 不确定度测定结果
综上所述, 可得用岛津GC2010PLUS 气相色谱仪测定菜籽油中溶剂残留的不确定度为:(8.73±1.2)mg·kg-1,k=2,p=95%。
3 结论
本文对气相色谱法测定菜籽油中溶剂残留结果进行不确定度评定,结果表明,标准曲线拟合过程引入的不确定度最大,其次是标准物质、回收率、重复性和标准系列溶液配制引入的不确定度,样品称量引入的不确定度最小。因此,在以后实验操作过程中,检验员可增加标准曲线溶液和平行样品的测定次数,保证标准物质的有效性,规范实验操作,减少随机误差,定期保养实验仪器来减小测量的不确定度,以提高检测结果的准确性和可信度。
猜你喜欢
口腔护理用品工业(2021年4期)2021-11-02
化工设计通讯(2020年10期)2020-09-17
中国油脂(2019年3期)2019-04-29
中成药(2018年6期)2018-07-11
中国粮油学报(2018年12期)2018-03-19
Cancer Biology & Medicine(2016年4期)2017-01-13
中国氯碱(2016年9期)2016-11-16
全面腐蚀控制(2016年11期)2016-02-14
中国粮油学报(2016年5期)2016-01-23
长江大学学报(自科版)(2014年27期)2014-02-27
