
一类受生物启发的双膦双硒镍配合物的合成及其电催化产氢性能
2021-04-02 02:23:52谢安潘中华骆耿耿
物理化学学报 2021年3期
谢安,潘中华,骆耿耿,3,*
1厦门理工学院材料科学与工程学院,福建省功能材料及应用重点实验室,福建 厦门 361024
2华侨大学材料科学与工程学院,福建 厦门 361021
3中国科学院福建物质结构研究所,结构化学国家重点实验室,福州 350002
1 引言
氢能具有高燃烧值和燃烧产物无污染等特点,是当前最有前途的清洁能源之一1,2。其中,电催化产氢被认为是可持续性生产氢气的一种有效手段3,4。因此,如何在分子水平上研制低成本的高效产氢催化剂则成为备受瞩目的研究课题3–5。自然界中,厌氧微生物体内存在一种能够催化质子还原的酶,即氢化酶6。根据其所含元素的组成及结构不同,氢化酶通常被分为[FeFe]氢化酶,[NiFe]氢化酶和[Fe]氢化酶(又称Hmd氢化酶)。其中的[NiFe]氢化酶具有催化可逆分裂重组分子氢的能力,因此对这类酶的活性中心结构,催化机理及化学模拟的研究一开始就引起科学家们的广泛关注6,7。
[NiFe]氢化酶的活性中心结构可描述为:由Ni和另外一个金属中心Fe组成的异核配合物,主要配位环境含4个半胱氨酸残基,其中2个半胱氨酸残基S作为端基与Ni配位,余下的2个半胱氨酸残基S除了与Ni配位外同时与Fe中心形成桥连。[NiFe]氢化酶旗下还有一类非常特殊的酶,即[NiFeSe]氢化酶8。[NiFeSe]氢化酶的活性中心仅仅是把与Ni配位的一个半胱氨基残基的S替换成Se原子。然而,这样简单的原子替换不仅提高了[NiFeSe]氢化酶的催化产氢效率,而且还赋予了[NiFeSe]氢化酶一些特殊的性质:如其在空气或氢气氛围下,催化产氢活性较少被抑制8。相对于[NiFe]氢化酶,[NiFeSe]氢化酶所表现出较高的催化活性跟其所取代的Se原子密切相关:通常认为,是因为[NiFeSe]氢化酶上的Se原子更容易被质子化有关9。[NiFeSe]氢化酶的特殊结构、性质及催化机制强烈激发科学家们投入设计并合成各种与[NiFeSe]氢化酶活性中心结构相似的镍铁硒或镍硒配合物(也即受生物启发的模拟物)9–11,以便深入理解这类氢化酶活性中心及催化机理,同时也为研制高效产氢活性提供一条仿生途径。
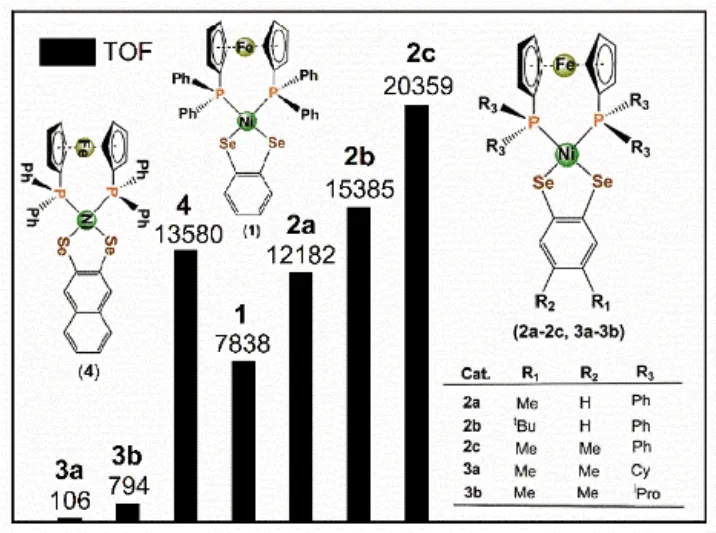
近年来,我们课题组在产氢分子催化剂方面做了一些探索12–17。其中,在开展[NiFeSe]氢化酶活性中心的化学模拟过程中,我们在2018年报道18了一例由1,2-苯二硒、1,1'-双(二苯膦)二茂铁与镍离子所配位形成的具有平面构型的镍硒配合物1(图1a),当测试其电化学还原质子催化活性时,该催化剂给出催化转化频率(TOF)为7838 s-1(以三氟乙酸为质子源),是类质同构的镍硫配合物的13倍,同时该镍硒配合物1还表现出较高的耐氧性。为进一步在分子水平上修饰调控这类镍硒配合物的催化产氢活性,在本工作中,我们分别尝试改变双硒和双膦配体上的取代基,一共合成表征了三个系列的双膦双硒镍配合物(2a–2c,3a–3b,4;图1b),详细研究双硒和双膦配体上取代基结构的不同对镍硒配合物催化产氢活性的影响。

图1 (a)先前课题组报道的镍硒配合物1的分子结构示意图;(b)本工作中新合成的双膦双硒镍配合物(2a–2c,3a–3b,4)的分子结构示意图Fig.1 (a) Molecular structure of the previous Ni(II) complex 1; (b) Molecular structures of newly synthesized Ni-based complexes (2a-2c, 3a-3b, 4) in this work.
2 实验部分
2.1 试剂与仪器
如未经特殊说明,实验中所使用的化学药品和试剂均为分析纯商品,使用前未进行纯化处理。所有对水和氧敏感的反应均按照标准的Schlenk真空技术操作,N2保护下进行。四氢呋喃使用前先用0.3 nm分子筛进行预处理。然后加入金属钠回流除水后,再蒸馏后使用。反应过程用薄层层析板(TLC)跟踪,紫外灯下检测。产物用硅胶柱层析分离。TLC用青岛海洋化工厂生产的GF-254硅胶板。柱色谱以硅胶200–300目为固定相。
核磁共振(NMR)采用Bruker PLUS 500 MHz核磁共振谱仪,以氘代氯仿(CDCl3)为溶剂,四甲基硅烷为内标。电化学循环伏安测试使用CHI 750E电化学工作站(上海辰华),测试采用三电极体系,以玻璃碳电极(直径3 mm)为工作电极,使用前用0.5 μm抛光剂打磨10 min;以饱和甘汞电极为参比电极,辅助电极为铂丝。用于循环伏安测试的乙腈(Aldrich,色谱纯),使用前用分子筛干燥后蒸馏。使用0.1 mol·L-1的四正丁基六氟磷酸铵(TBAPF6)的乙腈溶液为电解液。测试前,向电解液中通入干燥的氩气,采用鼓泡方式除氧10 min,并且保持测试在氩气氛围下进行。
2.2 双膦双硒镍配合物(2a–2c,3a–3b,4)的合成
不同取代基修饰的双膦双硒镍配合物的合成参照文献18。其中,聚3,4-二硒甲苯,聚1,2-二硒-4-叔丁基苯,聚1,2-二硒-4,5-二甲苯,和聚2,3-二硒萘的合成分别参照文献19进行。双膦双硒镍配合物的具体合成过程描述如下:
配合物2a的合成:在氩气保护下,往100 mL圆底烧瓶中加入1,1’-双(二苯基膦)(0.166 g,0.3 mmol)和干燥的四氢呋喃(10 mL),随后滴加双(三苯基膦)氯化镍(0.166 g,0.3 mmol)和甲醇(10 mL)。室温下搅拌反应5 h后,旋蒸除去溶剂,固体残渣重新溶解于二氯甲烷(10 mL)中得到暗绿色的溶液。将聚3,4-二硒甲苯(0.0795 g,0.3 mmol)与硼氢化钠(0.0456 g,1.2 mmol)加入甲醇(5 mL)中,然后缓慢滴加到上述暗绿色溶液,得到棕色溶液,继续搅拌反应50 min,旋去溶剂,粗产物用硅胶柱提纯(淋洗剂:Vhexane:Vdichloromethane:Vethylacetate= 2 : 1 :1),得到目标产物2a,产率36%。1H NMR (500 MHz,CDCl3):δ7.94 (m,8H)、7.48 (t,4H)、7.35(t,8H)、7.20 (d,1H)、7.16 (s,1H)、6.68 (dd,1H)、4.36 (s,4H)、4.24 (s,4H)、2.17 (s,3H)。31P NMR (500 MHz,CDCl3):δ27.41。
配合物2b的合成:与配合物2a的合成流程类似,但用聚1,2-二硒-4-叔丁基苯代替聚3,4-二硒甲苯,产率27%。1H NMR (500 MHz,CDCl3):δ7.94(m,8H)、7.47 (t,4H)、7.37 (m,9H)、7.23 (dd,1H)、6.90 (d,1H)、4.35 (s,4H)、4.24 (s,4H)、1.21 (s,9H)。31P NMR (500 MHz,CDCl3):δ27.39。
配合物2c的合成:与配合物2a的合成流程类似,但用聚1,2-二硒-4,5-二甲苯代替聚3,4-二硒甲苯,产率17%。1H NMR (500 MHz,CDCl3):δ7.91(m,8H)、7.46 (t,4H)、7.33 (t,8H)、7.11 (s,2H)、4.35 (s,4H)、4.25 (s,4H)、2.08 (s,6H)。31P NMR(500 MHz,CDCl3):δ27.33。
配合物3a的合成:与配合物2c的合成流程类似,但用1,1'-二(二环己膦)二茂铁代替1,1'-双(二苯膦)二茂铁,产率20%。1H NMR (500 MHz,CDCl3):δ7.42 (s,2H)、4.39 (t,4H)、4.38 (t,4H)、2.22(s,6H)、2.10–1.10 (m,44H)。31P NMR (500 MHz,CDCl3):δ26.19。
配合物3b的合成:与配合物2c的合成流程类似,但用1,1'-双(二异丙膦)二茂铁代替1,1'-双(二苯膦)二茂铁,产率24%。1H NMR (500 MHz,CDCl3):δ7.42 (s,2H)、4.46 (t,4H)、4.42 (t,4H)、2.64(m,4H)、2.21 (s,6H)、1.37 (m,24H)。31P NMR(500 MHz,CDCl3):δ36.96。
配合物4的合成:与配合物2a的合成流程类似,但用聚2,3-二硒萘代替聚3,4-二硒甲苯,产率41%。1H NMR (500 MHz,CDCl3):δ7.98 (m,8H)、7.80 (s,2H)、7.54 (m,2H)、7.49 (t,4H)、7.38 (t,8H)、7.20 (m,2H)、4.37 (s,4H)、4.27 (s,4H)。31P NMR (500 MHz,CDCl3):δ27.65。
2.3 单晶结构测试
配合物4的晶体结构测定采用日本理学RAXIS RAPID Image Plate单晶衍射仪。入射光源为石墨单色器单色化的Mo-Kα射线(λ= 0.071073 nm),衍射点收集方式采用ω扫描。采用ABSCOR软件20对数据进行对称等效反射吸收校正,选择了最为可能的空间群,运用SHELXS-97软件21采用直接法完成结构解析,通过SHELXL-97软件22采用F2全矩阵最小二乘法对结构进行精修,非氢原子位置直接从差分F-图中找出,然后用最小二乘法并做各向异性精修。氢原子坐标采用理论加氢来确定。配合物4的晶体CCDC号为1961412。
2.4 电催化产氢性能测试
电催化产氢性能测试条件参考先前文献18:将镍硒配合物溶解在0.1 mol·L-1的TBAPF6的乙腈溶液中进行循环伏安测试,循环伏安测试需要在静止状态进行测定,以保证测试是扩散控制过程;然后将准备好的溶液以及三个电极至于测量的容器中,用高纯氩气鼓泡以除去溶液中存在的氧气,设置扫描速率0.1 V·s-1,随后将质子酸添加到测试溶液中,记录循环伏安曲线图,重复添加数次以增加质子酸的浓度,记录每次添加酸后的循环伏安曲线图。为了保证工作电极面积的一致性及实验结果良好的重现性,在每次使用前,工作电极均用三氧化二铝粉末打磨抛光,并在蒸馏水中超声10 min,然后用有机溶剂丙酮冲洗干净,直到电极表面光滑平整。通过气相色谱仪(岛津GC-2014C)分析检测所取出气体中氢气的峰面积,检测器为热岛检测器及0.5 nm分子筛。氢气量可参考文献7,23通过气相色谱检测到氢气的峰面积,然后利用外标曲线获得。
3 结果与讨论
3.1 双膦双硒镍配合物的合成及表征
参考先前文献方法18,双膦双硒镍配合物的合成可分为两步(如图2所示):首先根据文献19合成得到聚邻苯二硒衍生物;然后利用硼氢化钠对聚邻苯二硒衍生物进行解聚得到单体的二硒钠盐,后与含二茂铁的双膦配体以及双(三苯基膦)氯化镍一步反应,经硅胶柱分离后得到空气中稳定的双膦双硒镍配合物(2a–2c,3a–3b,4)。为了进一步研究确定这些配合物的立体结构,我们利用扩散法培养了镍硒配合物4的单晶,并利用X射线衍射技术测定了该化合物的单晶结构。单晶结构分析表明,配合物4的晶体属单斜晶系C2/c,,其结构中含有一个溶剂分子CH3CN。从图3所示的分子椭球图可以看出,该配合物中金属Ni分别与1,2-萘二硒的两个Se原子,双(二苯膦)二茂铁中的两个P原子进行螯合配位,呈现平面四边形构型。配合物中Ni-Se的平均键长是0.2293 nm,稍短于[NiFeSe]氢化酶中Ni-Se的平均键长(0.244 nm)24,但和平面结构的双硒镍配合物[(C4H9)4N]2[Ni(bds)2]中的Ni–Se距离(0.2259 nm)19及已报道的配合物1的Ni–Se距离(0.2292 nm)18相当。另外,分子内NiII和FeII之间的距离为0.4217 nm,可排除Ni-Fe的金属键合作用。

图2 双膦双硒镍配合物(2a–2c,3a–3b,4)的合成路线Fig.2 Synthetic routes of complexes (2a–2c, 3a–3b, 4).

图3 双膦双硒镍配合物4的分子椭球图Fig.3 The ORTEP of complex 4 with the ellipsoids drawn at 40% probability level.
3.2 双膦双硒镍配合物的氧化还原性质
为了研究配体取代对这类双膦双硒镍配合物的氧化还原电位的影响,以TBAPF6为支持电解质,在除气的CH3CN溶液中分别测量了配合物2a–2c,3a–3b和4的氧化还原电位,测试过程中电位扫描方向为负方向,扫描速率为0.1 V·s-1,所有电位均相对于二茂铁的半波电位。循环伏安曲线和相应的电化学数据分别列于图4和表1。从图4和表1的结果可知:(1)双膦双硒镍配合物2a–2c,3a–3b和4在负电位区-1.5 – -1.8 V范围内均表现出一个可逆的还原过程,且ipc/ipa≈ 1,峰位差分别为63–68 mV,可归属于金属中心NiII→NiI的单电子还原;在正电位区0.1–0.2 V范围内表现出不可逆的氧化过程,归属于FeII→FeIII的单电子氧化。(2)配合物2a–2c的NiII/NiI单电子还原峰电位与先前所报道的配合物1相比,随着双硒配体取代基给电子效应存在差异,会导致中心镍原子电子云密度不同,表现出还原峰电位出现负移;配合物4的NiII/NiI还原峰电位与配合物1相比则正移了约40 mV,可能是由于硒配体共轭度的提高有利于降低配合物中的NiII/NiI还原峰电位;配合物3a–3b的NiII/NiI还原峰电位与配合物1相比偏差较大,分别负移了223和108 mV,归因于磷原子上的苯环被环己烷和异丙基所替代。

图4 双膦双硒镍配合物2a–2c,3a–3b和4的循环伏安曲线Fig.4 CVs of complexes (2a-2c, 3a-3b, 4).

表1 镍配合物2a–2c,3a–3b和4的氧化还原电势aTable 1 Electrochemical redox data for the nickel-based complexes (2a–2c, 3a–3b,4 ) a.
3.3 电催化产氢性能测试
在电催化产氢体系中,调控分子催化剂的结构在很大程度上会影响体系的催化产氢活性3,4。考虑到先前文献18所报道的由1,2-苯二硒和1,1'-双(二苯膦)二茂铁所配位形成的配合物1的电催化活性为TOF = 7838 s-1,我们在此基础上,对配合物(2a–2c,3a–3b和4)在三氟乙酸(TFA)存在下进行电化学测试,分别研究双硒配体上不同取代基,以及双膦配体上不同取代基等不同的结构修饰方式对催化产氢活性的影响,其中的催化转化频率TOF根据参考文献7,14,18进行计算。
3.3.1 双硒配体上取代基不同对体系电催化性能的影响
我们首先考察双硒配体上取代基的给电子基团对分子催化剂产氢性能的影响,鉴于配合物2a–2c在CH3CN溶液中的还原电位及文献报道18。图5a–c给出了在2a–2c在0.1 mol·L-1TBAPF6/CH3CN溶液中加TFA后的循环伏安叠加图。由图可知,随着TFA浓度的增加,镍配合物2a–2c均在NiII还原峰电流逐渐增长,且向阴极方向有规律的移动,同时相应的氧化峰消失;这是典型的电化学催化质子还原产氢行为的特征现象7,18;催化过程中从电极表面均可以看到明显气泡生成,同时气相色谱检测到气体为氢气,证实这些配合物在-1.5 – -1.7 V处的还原峰具有电催化质子还原的活性。当滴加的TFA浓度> 75 mmol·L-1时,所对应的-1.5 – -1.7 V处的还原峰基本不在升高,意味着这些配合物的催化性能已达到饱和。由于这些镍硒配合物的双膦配体都是1,1'-双(二苯膦)二茂铁,不同在于双硒配体上取代基的改变,这样,在相同配合物浓度及在同样的TFA酸浓度(75 mmol·L-1)的条件下,这些配合物的电催化活性的表现为:双硒配体上含两个甲基取代的配合物2c的电催化活性最高(TOF =20359 s-1),其次是双硒配体上含一个叔丁基取代的配合物2b (TOF = 15385 s-1),而双硒配体上仅含一个甲基取代的配合物2a的催化活性最低(TOF =12182 s-1)。比较配合物2a–2c我们可以看出,含1,2-二硒-4,5-二甲基的配合物2a要比含其他两种双硒配体的电化学还原质子催化活性效果高。值得一提的是,这类镍硒配合物中,配体给电子基团的增加对分子催化剂催化产氢性能影响与2016年吴屹影课题组25报道的钼硫配合物具有相似性。另外,正如图5d所示,这些镍硒配合物2a–2c的TOF值都已高于先前我们所报道的配合物1 (TOF =7838 s-1)18。

图5 双膦双硒镍配合物2a–2c的催化TFA还原产氢的电化学行为Fig.5 CVs of 2a–2c with varying amount of TFA in 0.1 mol·L-1 TBAPF6/CH3CN at a scan rate of 0.1 V·s-1.
3.3.2 双膦配体上不同取代基对体系电催化性能的影响
为了考察双膦配体上不同取代基对分子催化剂产氢性能的影响,我们合成了双膦配体上为环己烷基团和异丙烷基团取代的配合物3a–3b,考虑到这两个配合物在CH3CN中的溶解度较差,我们参照文献25中所使用的溶剂N,N'-二甲基甲酰胺(DMF),以TFA为质子源对在配合物3a–3b进行循环伏安加酸实验,来验证其催化质子还原产氢能力。图6给出了在0.1 mol·L-1TBAPF6/DMF溶液中加TFA后的循环伏安叠加图。由图可知,随着TFA浓度的增加,配合物3a在NiII还原峰附近的的阴极电流也出现明显的增加,同时相应的氧化峰消失,这是典型的电化学催化质子还原产氢行为;当TFA浓度仅为5 mmol·L-1时,膦配体上环己烷基团取代的配合物3a的催化能力达到最大值,对应的TOF值为106 s-1;相比之下,膦配体上异丙烷基团取代的镍配合物3b的TOF最高可达794 s-1,是3a的7倍。然而,3a–3b的催化活性都远小于镍配合物1和2a–2c,表明膦配体上的取代基对配合物的催化性能影响很大:膦配体上苯环取代基相比于脂肪族如环己烷基团或异丙烷基团更有利于镍配合物的催化产氢反应的进行。
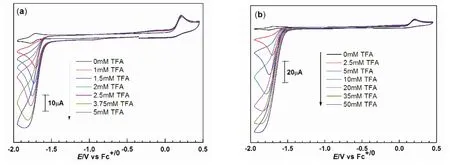
图6 双膦双硒镍配合物3a–3b的催化TFA还原产氢的电化学行为Fig.6 CVs of 3a–3b with varying amount of TFA in 0.1 mol·L-1 TBAPF6/DMF at a scan rate of 0.1 V·s-1.
3.3.3 双硒配体上共轭程度对体系电催化性能的影响
为了进一步考察双硒配体上共轭程度对分子催化剂产氢性能的影响,使用相同的双膦配体(即1,1'-双(二苯膦)二茂铁),但把配合物1中的1,2-苯二硒换成1,2-萘二硒合成得到新的镍硒配合物4。向溶有0.25 mmol·L-1配合物4的电解池中连续加入质子酸TFA,并通过循环伏安法监测还原电位及电流强度的变化。如图7a所示,随着配合物4的CH3CN溶液中TFA浓度不断增加,NiII还原峰-1.48 V附近的阴极电流也出现明显的增加,同时相应的氧化峰消失,在电极的表面逐渐有气泡逸出,色谱检测确认氢气的生成。当TFA浓度高达75 mmol·L-1时,质子还原的起始电位达到-1.62 V,此时,配合物4催化转化频率可达13580 s-1,与同等条件下配合物1 (TOF = 7838 s-1)相比18,催化活性明显增强,表明提高双硒配体的共轭程度有助于增强此类分子催化剂的催化产氢活性。此外,为了进一步确认这类镍硒分子催化剂的催化产氢稳定性,根据参考文献7,18,我们对配合物4进行了加酸控制电位电解实验,如图7b所示,配合物4在75 mmol·L-1TFA存在下电解一个半小时左右,持续有电量通过,暗示该均相体系的分子催化剂在催化过程能够维持一定的稳定性。

图7 (a) 双膦双硒镍配合物4的催化TFA还原产氢的电化学行为(b)配合物4在75 mmol·L-1 TFA中的控制电位电解曲线Fig.7 (a) CVs of 4 with varying amount of TFA in 0.1 mol·L-1 TBAPF6/CH3CN at a scan rate of 0.1·V s-1;(b) Charge buildup of complex 4 versus the applied potential (-1.5 V).Data has been deducted blank.
4 结论
本工作中,我们通过配体取代方式设计合成了六个双膦双硒镍配合物(2a–2c,3a–3b,4)作为[NiFeSe]氢化酶的模型配合物。以这些镍硒配合物为分子催化剂,TFA为质子源,研究了其电催化产氢行为,探索了双硒配体上的不同取代基,以及双膦配体上不同取代基等结构修饰方式对催化产氢活性的影响。结果表明:(1)当双膦配体不变,随着双硒苯配体上的取代基个数的增加,相应的分子催化剂的催化活性不断增强;若不改变双膦配体,而将双硒苯配体替换成共轭度更高的双硒萘配体后,相应的分子催化剂的催化活性也不断增强;这说明增加双硒配体的给电子基团能力或提高双硒配体的共轭度可显著提高镍硒配合物的催化产氢活性。(2)当双硒配体不变,而将双膦配体中磷原子上苯基换成环己基或异丙基后,相应的分子催化剂的溶解度和催化活性则大幅度降低。这表明双膦配体中用非苯基取代基后会大大降低此类镍硒配合物的催化产氢能力。
尽管这类镍硒配合物电催化产氢的TOF值已优于天然氢化酶,但考虑到这些镍硒配合物的过电势(> 600 mV)仍然较高,今后我们的研究工作将集中在对镍硒配合物进行结构修饰(如引进类似Dubois的悬挂键配体),以期降低这类分子催化剂的过电势,以期得到高效稳定的电催化产氢分子催化剂。
猜你喜欢
肿瘤防治研究(2023年2期)2023-04-05 11:02:25
国际放射医学核医学杂志(2020年2期)2020-05-30 12:39:52
中国有色金属学报(2018年2期)2018-03-26 07:58:37
无机盐工业(2017年5期)2017-05-25 00:37:34
郑州大学学报(理学版)(2017年1期)2017-04-07 01:09:39
化工管理(2017年25期)2017-03-05 23:32:36
中南大学学报(自然科学版)(2016年2期)2017-01-19 07:37:25
中国资源综合利用(2016年7期)2016-02-03 03:00:13
中国药业(2014年17期)2014-05-26 09:08:04
影像科学与光化学(2014年5期)2014-03-11 16:03:22

- 物理化学学报的其它文章
- SnO2表面卤化提高钙钛矿太阳能电池光伏性能
- ZrO2包覆高镍LiNi0.8Co0.1Mn0.1O2正极材料提高其循环稳定性的作用机理
- Regulating Electron Transport Band Gaps of Bovine Serum Albumin by Binding Hemin
- 基于血红素衍生的中空非贵金属催化剂氧还原反应电催化活性
- Influence of NaOH Concentration on Sodium Storage Performance of Na0.44MnO2
- Photocathodic Protection on Stainless Steel by Heterostructured NiO/TiO2 Nanotube Array Film with Charge Storage Capability