
基于血红素衍生的中空非贵金属催化剂氧还原反应电催化活性
2021-04-02 02:23:52李琳沈水云魏光华章俊良
物理化学学报 2021年3期
李琳,沈水云,魏光华,章俊良,3,*
1上海交通大学机械与动力工程学院燃料电池研究所,上海 200240
2上海交通大学巴黎高科学院,上海 200240
3上海交通大学动力机械与工程教育部重点实验室,上海 200240

1 引言
燃料电池因具有能量转化效率高、清洁、无污染等优点被视为新能源汽车的终极解决方案,成为当前研究的热点。然而,其阴极氧还原反应动力学非常缓慢,相比于阳极的氢气氧化反应低了5个数量级1,从而严重制约了电池的整体性能。贵金属铂及其过渡金属(Co、Ni、Fe等)合金类催化剂对于氧还原反应具有很好的催化活性,然而铂资源短缺、价格昂贵,大大提高了燃料电池的成本,限制了其大规模的应用。因此,寻找价格低廉、性能良好的非贵金属氧还原电催化剂成为近年来的研究热点2–4。
非贵金属催化剂主要由过渡金属(Fe、Co、Mn等)、氮和碳组成,而金属大环化合物可以独立承担金属源、氮源和碳源作用,是一种理想的非贵金属氧还原电催化剂前驱体。1964年Jasinski5首次将金属大环化合物酞菁钴(CoPc)用作氧还原催化剂并表现出良好的电催化活性,随后Lalande等6发现经过进一步高温热处理可以有效提高金属大环化合物的氧还原催化性能。大环化合物用于制备非贵金属氧还原催化剂引起众多研究者的关注7–10。He等11将Cu酞菁与Fe酞菁负载在碳载体上经过热处理得到CuFe/C非贵金属催化剂,该催化剂在0.1 mol·L-1KOH溶液中0.8 V下的活性为24.5 mA·cm-2,高于Pt/C催化剂的21.4 mA·cm-2。
血红素作为一种天然的同时含有Fe、N、C的金属大环化合物也广泛应用于制备非贵金属氧还原电催化剂12–15。Jiang等16使用XC72R作为载体,氯化血红素与钴卟啉作为前驱体合成CoFeNx/C催化剂,探究其在酸性和碱性介质中对氧还原反应的催化活性,发现氧还原反应动力学速率在碱性介质中比在酸性介质中高4倍。Jiang等17采用高比表面的石墨烯纳米片作为碳载体负载血红素,同时进行超声处理使催化剂尺寸变小,将比表面积提高至未经过超声处理催化剂的2.7倍,该催化剂在0.5 mol·L-1H2SO4溶液中氧还原反应的半波电位达到0.75 V,制备的氢氧燃料电池的峰值功率密度达到了300 mW·cm-2。Xie等18使用盐酸处理羟基化血红素使其质子化,得到自组装的血红素,碳化后产生纳米孔,该多孔催化剂相较于直接用血红素制备得到的微米尺度催化剂具有更大的比表面积,可以暴露更多的活性位点,从而提高氧还原性能。
为了提高非贵金属催化剂的氧还原电催化活性,不仅需要设计化学组分,还需要调控其结构包括形貌、颗粒尺寸、孔道结构等,两者均对氧还原性能有重大的影响19–22。合理的结构设计需要实现1)提高活性位点密度,通过调控催化剂形貌等方式增加暴露的活性位点,高密度的催化活性位点有利于减小动力学阻力;2)构建多孔结构,既可用于活性位点的负载,也可用于氧气与电解液的传输与扩散,构建有效的氧还原反应三相(固-液-气)区域,减小传质阻力23。中空结构的设计可以有效提高非贵金属电催化剂的氧还原活性,一方面其催化活性位可以同时分散在内表面和外表面24,大大提高活性位点密度,同时催化剂中存在大量介孔/微孔,加强了活性位点与反应物之间的接触,有利于反应的进行。
模板法是制备中空结构常用的方法之一,它是通过物理或化学方法将相关材料沉积到模板表面而后移除模板,从而得到具有模板形貌与尺寸的纳米材料的过程。Cheon等25采用有序二氧化硅作模板,将前驱体研磨煅烧,然后用氢氟酸刻蚀二氧化硅模板得到有序介孔催化剂FeCo-OMPC,在0.9 V下的动力学电流为2.44 mA·cm-2,比Pt/C催化剂(1.76 mA·cm-2)高39%,同时表现出更好的稳定性和耐甲醇性。Tan等26基于水热法,以锰氧化物为模板,表面包覆聚合物,然后煅烧碳化,酸洗去除氧化物模板后得到具有大量介孔的催化剂MnO-m-N-C,该催化剂在0.1 mol·L-1KOH溶液中半波电位达0.81 V,相比于MnO催化剂(0.65 V)有大幅度提升。然而,使用硬模板需要采用强酸或强碱去除模板,不仅会污染环境,还会造成部分活性位点的损失,软模板则通常需要使用有机化合物,成本较为昂贵。
因此,本文提出采用血红素作为前驱体,以易被去除的氯化钠为模板制备具有中空形貌的催化剂,以期提高催化剂的比表面积,从而改善其氧还原性能。
2 实验部分
2.1 Hemin-HD和Hemin-D催化剂的制备
称取0.2 g氯化血红素(C34H32ClN4O4Fe,95%,阿拉丁)溶解于10 mLN,N-二甲基甲酰胺(HCON(CH3)2,≥99.8%,Sigma-Aldrich)中形成溶液A。称取2 g氯化钠(NaCl,≥99.5%,国药)溶解于40 mL超纯水中形成溶液B。在搅拌条件下将溶液B缓慢加入至溶液A中,在160 ℃油浴下搅拌蒸干溶剂,60 ℃真空干燥8 h,所得固体研磨至粉末备用。将得到的前驱体置于刚玉舟中,在氮气氛围保护下900 ℃高温处理3 h,升温速率为5 ℃·min-1。冷却至室温后,将得到的样品用大量去离子水洗涤去除氯化钠模板,得到最终的催化剂,标记为Hemin-HD (Hemin hollow derivative)。
作为对照实验,未使用氯化钠作为模板剂,在相同条件下热解氯化血红素得到的催化剂标记为Hemin-D (Hemin derivative)。具体制备方法如下:直接称取0.2 g氯化血红素在氮气氛围保护下在900 ℃煅烧3 h,得到最终的Hemin-D催化剂。
2.2 催化剂的物理化学表征
采用JEM-2100F型透射电子显微镜(TEM,JEOL,日本)观测Hemin-HD和Hemin-D催化剂的微观形貌;采用D8 ADVANCE DA VINCI型多功能X射线衍射仪(XRD,Bruker,德国)表征其晶体结构,射线源采用CuKα辐射,波长为0.15405 nm,测试范围为20°–80°;采用3H-2000PS1型比表面及孔径分析仪(贝士德仪器,中国)测定Hemin-HD和Hemin-D催化剂的氮气吸脱附等温线及孔径分布;采用Kratos AXIS Ultra DLD型X射线光电子能谱仪表征其表面的元素组成及化学态。
2.3 催化剂的电化学测试
Hemin-HD和Hemin-D催化剂的氧还原反应活性采用CHI760e电化学工作站(上海辰华仪器有限公司,中国),在三电极体系下进行评价。以负载有催化剂的玻碳电极为工作电极,饱和甘汞电极为参比电极,铂片为对电极,电解质为0.1 mol·L-1KOH溶液。工作电极的制备方法为:称取5 mg非贵金属催化剂,分散在0.5 mL 0.25% (w)的Nafion溶液中,超声30 min得到催化剂浆料,取12 μL催化剂浆料滴于玻碳电极表面,干燥得到工作电极,非贵金属催化剂的载量为0.6 mg·cm-2。为做对照,制备了商业Pt/C催化剂的工作电极,Pt载量为30 μg·cm-2。循环伏安测试和旋转圆盘电极测试在氧气饱和的0.1 mol·L-1KOH溶液中进行,扫描速度5 mV·s-1。
3 结果与讨论
图1所示为Hemin-HD催化剂的制备过程示意图。氯化血红素和氯化钠溶解在溶液中,随着温度的升高,溶剂逐渐挥发,氯化钠开始结晶,由于氯化血红素与氯化钠的离子间相互作用,氯化血红素包覆在氯化钠表面27,溶剂完全挥发后,得到氯化血红素包裹氯化钠的前驱体。在热处理过程中,氯化血红素发生碳化但仍然包覆在氯化钠表面,最后采用去离子水洗涤,去除氯化钠模板,得到中空的Hemin-HD催化剂。
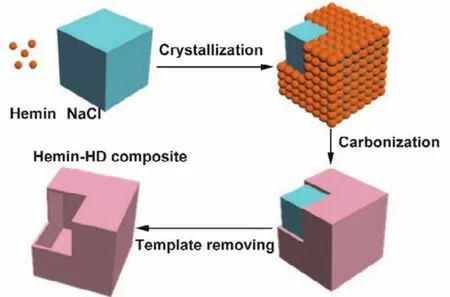
图1 Hemin-HD催化剂的制备过程示意图Fig.1 Schematic of the fabrication process of the Hemin-HD electrocatalyst.
图2a,b分别为制备的Hemin-D和Hemin-HD催化剂的TEM图,由图可见,两者的形貌结构存在明显差异。Hemin-D催化剂表现为多孔碳负载铁或铁氧化物颗粒的结构,这可能是由于氯化血红素作为含碳有机物经过高温碳化后生成石墨碳,而其中含有的铁元素经过热处理后生成铁单质或铁氧化物颗粒。Hemin-HD催化剂表现出了中空结构,这是由于在前驱体生长过程中,氯化血红素包覆在氯化钠晶体上,将氯化钠模板去除后,则催化剂表现出中空形貌,说明仅用去离子水洗涤就可有效去除氯化钠模板。

图2 (a) Hemin-D和(b) Hemin-HD的透射电镜图Fig.2 TEM images of (a) Hemin-D and (b) Hemin-HD.
图3为Hemin-D和Hemin-HD催化剂的XRD谱图,由图可见,两者表现出的晶相物种基本一致。两种催化剂均在26°位置出现C(002)的特征峰,在45°位置出现Fe (PDF #06-0696)的特征峰,还表现出了Fe3O4(PDF #65-3107)的特征峰,说明催化剂中除了石墨碳,主要存在的晶相物种为Fe和Fe3O4。此外,Hemin-HD催化剂的XRD谱图中未出现其他晶相物种的特征衍射峰,说明NaCl模板在去离子水洗涤过程中已被完全去除。

图3 Hemin-D和Hemin-HD催化剂的XRD谱图Fig.3 XRD patterns of Hemin-D and Hemin-HD catalysts.
图4a为获得的Hemin-D和Hemin-HD催化剂的氮气吸脱附曲线,由此可以得到催化剂的比表面积和孔隙分布等信息。通过Brunner-Emmet-Teller(BET)多点法计算可知,Hemin-HD催化剂具有较大的比表面积,为857.6 m2·g-1,是Hemin-D催化剂比表面积(114.7 m2·g-1)的7.5倍,这是由于NaCl模板的使用,可以有效分散前驱体,同时构建了中空的结构,从而提高了催化剂的比表面积。图4b为Hemin-D和Hemin-HD催化剂的孔径分析,由图可见,Hemin-HD催化剂中含有大量的微孔和介孔。Hemin-HD催化剂中的孔体积(1.05 mL·g-1)明显高于Hemin-D催化剂(0.22 mL·g-1),总孔体积是Hemin-D催化剂的4.8倍,说明采用氯化钠模板法制备催化剂可显著提高材料的比表面积及孔体积。众多文献研究表明,高比表面积和丰富的微孔或介孔结构一方面可以增加暴露的活性位点,提高利用率22,28,另一方面可以促进催化剂与氧气的充分接触29,从而进一步改善Hemin-HD催化剂的氧还原性能。

图4 Hemin-D和Hemin-HD催化剂的(a)氮气吸脱附曲线和(b)孔径分布曲线Fig.4 (a) N2 adsorption-desorption isotherms and(b) pore size distribution curves of Hemin-D and Hemin-HD catalysts.
为了探究Hemin-D和Hemin-HD催化剂的表面元素组成,我们对催化剂进行了XPS分析。图5分别展示了两种催化剂的XPS全谱,以及C 1s、N 1s和Fe 2p的高分辨XPS谱图。由图可见,两种催化剂主要由C、N、O、Fe元素组成。其中Hemin-D催化剂中N和Fe的相对原子含量分别为1.46%和0.68%,Hemin-HD催化剂中N和Fe的相对原子含量分别为1.68%和3.72%。Hemin-HD催化剂表面的Fe含量明显高于Hemin-D催化剂,这可能是因为氯化钠的存在有效抑制了铁和铁氧化物颗粒的聚集,使得元素分布均匀,且其碳层厚度较小,而Hemin-D催化剂中元素分布不均匀,并且铁和铁氧化物颗粒可能被碳层包裹,超出了XPS的检测深度,故暴露出的Fe含量较少。Hemin-HD催化剂表面的N含量也高于Hemin-D催化剂,众多文献表明,Fe和N均对氧还原活性有重要的影响30–32,Hemin-HD催化剂表面的Fe和N含量较高,说明其表面活性组分较多,有利于提高氧还原反应活性。此外,进一步对N元素进行分峰,Hemin-D催化剂分为吡啶氮(N1,32.5%)、吡咯氮(N2,27.0%)和石墨氮(N3,40.5%),Hemin-HD催化剂分为吡啶氮(N1,36.4%)、吡咯氮(N2,21.8%)和石墨氮(N3,41.8%)。其中吡啶氮和石墨氮被认为对氧还原活性有促进作用33,而Hemin-HD催化剂中的吡啶氮和石墨氮含量均高于Hemin-D催化剂。
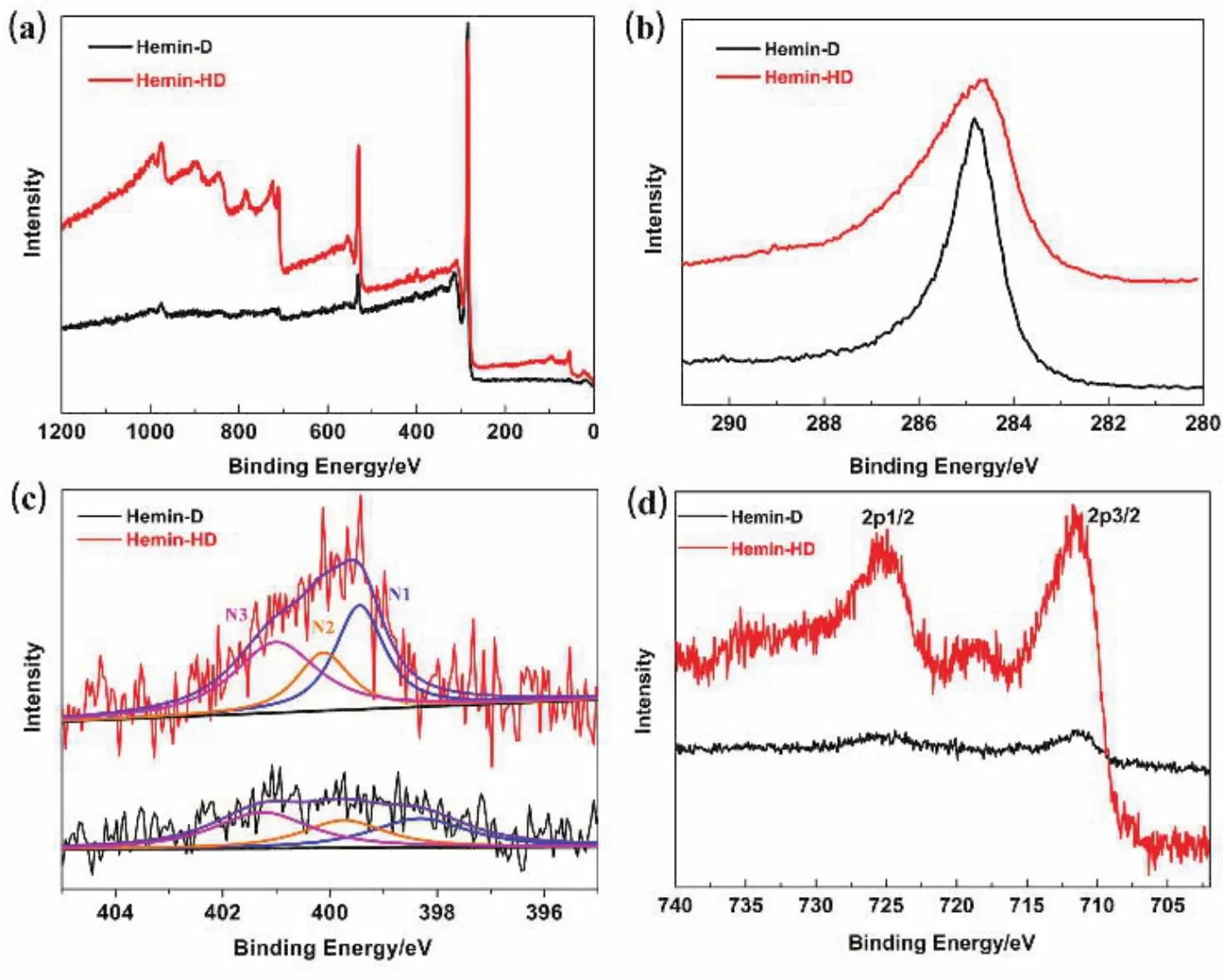
图5 Hemin-D和Hemin-HD催化剂的(a) XPS谱图,(b) C 1s,(c) N 1s和(d) Fe 2p谱图Fig.5 (a) XPS spectra, (b) C 1s, (c) N 1s, (d) Fe 2p of Hemin-D and Hemin-HD catalysts.
为了表征Hemin-D和Hemin-HD催化剂的氧还原活性,采用循环伏安法和旋转圆盘电极法对催化剂进行测试。图6a–c为两种催化剂分别在氧气饱和的0.1 mol·L-1KOH溶液中的循环伏安曲线和1600 r·min-1转速下的极化曲线对比,以及在1600 r·min-1转速下的极化曲线对应的塔菲尔曲线。由图可见,Hemin-HD催化剂在循环伏安曲线中的氧还原峰电位为0.87 V,比Hemin-D催化剂的氧还原峰电位高20 mV,在旋转圆盘测试中1600 r·min-1转速下的半波电位为0.82 V,比Hemin-D催化剂高20 mV,两种氧还原活性评价手段均表明,相对于Hemin-D催化剂,Hemin-HD催化剂表现出更高的氧还原催化活性。Hemin-D和Hemin-HD催化剂的塔菲尔斜率分别为86.8和81.2 mV·dec-1,塔菲尔斜率越低,反应动力学越快,说明Hemin-HD催化剂在0.1 mol·L-1KOH溶液中针对氧还原反应具有更快的反应动力学。图6d为Hemin-HD催化剂和商业Pt/C催化剂在1600 r·min-1转速下的极化曲线对比,由图可见Hemin-HD催化剂表现出更高的氧还原起始电位和相近的半波电位,这表明了Hemin-HD催化剂具有与Pt/C催化剂相当的氧还原催化活性。综合上述物理化学表征和电化学性能测试可以看出,采用氯化钠模板法制备催化剂可有效提高氧还原催化活性,其原因可能在于采用模板法形成的催化剂为中空结构,提高了催化剂的比表面积和孔体积,使得更多活性位点暴露在表面,同时改善了氧还原反应中三相界面的离子扩散和氧气传质速率,促进氧还原反应的进行。

图6 Hemin-D和Hemin-HD催化剂在0.1 mol·L-1 KOH溶液中的(a)循环伏安曲线,(b) 1600 r·min-1转速下线性扫描曲线和(c)塔菲尔曲线,(d) Hemin-HD催化剂和Pt/C催化剂在1600 r·min-1转速下的线性扫描曲线Fig.6 (a) CV curves, (b) polarization curves at 1600 r·min-1, (c) Tafel plots on Hemin-D and Hemin-HD and(d) polarization curves at 1600 r·min-1 on Hemin-HD and commercial Pt/C catalysts in 0.1 mol·L-1 KOH solution.
氧还原反应主要有两种途径:二电子反应途径和四电子反应途径,其中二电子反应途径的最终产物为双氧水,四电子反应途径的最终产物为水。对Hemin-D和Hemin-HD催化剂进行不同转速下的线性扫描,通过极化曲线在不同电位下的电流拟合得到相应的Koutecky-Levich (K-L)曲线。通过K-L方程可以计算电子转移数34,35:
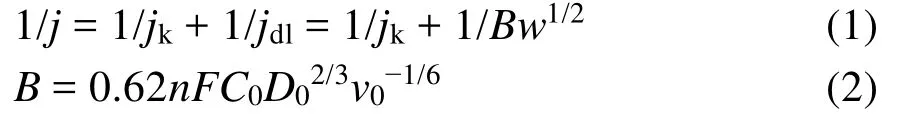
公式(1)中,j是测试得到的电流密度,jk是动力学电流密度,jdl是极限扩散电流密度,w是旋转速度,公式(2)中,n是电子转移数,F是法拉第常数,C0是氧气在电解质中的溶解度,D0是氧气在电解质中的扩散系数,v0是电解质的动力学粘度。
图7为Hemin-D和Hemin-HD催化剂在0.1 mol·L-1KOH溶液中不同转速下的旋转圆盘曲线及相应的K-L曲线,通过计算得到Hemin-D和Hemin-HD催化剂的电子转移数分别为3.7和3.6,说明两种催化剂对氧还原反应的催化主要以四电子途径进行。
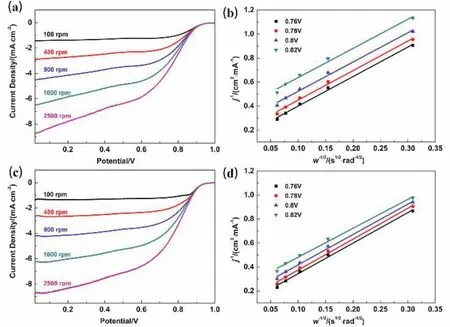
图7 (a, b) Hemin-D和(c, d) Hemin-HD催化剂在氧气饱和的0.1 mol·L-1 KOH溶液中不同转速下的线性扫描曲线及其相应的K-L曲线Fig.7 LSV curves in O2-satuarated 0.1 mol·L-1 KOH solution at different rotation rates and their corresponding K-L plots of (a, b) Hemin-D and (c, d) Hemin-HD catalysts.
4 结论
本文采用氯化钠作为模板,以血红素为前驱体设计合成了一种中空的铁基非贵金属催化剂Hemin-HD。该催化剂在碱性介质中表现出良好的氧还原催化活性,半波电位达0.82 V,与Pt/C催化剂相当。这主要是由于通过模板法制备得到中空结构催化剂,活性位可同时分散于内表面和外表面,从而暴露更多的活性位点,提高了催化剂的比表面积。另一方面催化剂中的孔道结构可以提高氧气传质速率,加强活性位点与反应物之间的接触,进一步促进氧还原反应的进行。相比于无模板的催化剂Hemin-D,Hemin-HD催化剂比表面积提升了6.5倍,孔容积增加了3.8倍,表现出更高的氧还原活性。
猜你喜欢
中学生数理化·中考版(2022年12期)2022-02-16 07:37:02
原子与分子物理学报(2020年5期)2020-03-17 07:00:16
奥秘(2016年12期)2016-12-17 15:48:57
爆笑show(2015年4期)2015-06-24 07:48:14
医学研究杂志(2015年11期)2015-06-10 06:44:03
淮南师范学院学报(2015年3期)2015-03-22 01:16:19
河北科技大学学报(2015年5期)2015-03-11 16:16:34
肝胆胰外科杂志(2015年2期)2015-02-27 11:11:49
无机化学学报(2014年4期)2014-02-28 17:31:23
应用技术学报(2014年1期)2014-02-28 14:52:11

- 物理化学学报的其它文章
- SnO2表面卤化提高钙钛矿太阳能电池光伏性能
- ZrO2包覆高镍LiNi0.8Co0.1Mn0.1O2正极材料提高其循环稳定性的作用机理
- Regulating Electron Transport Band Gaps of Bovine Serum Albumin by Binding Hemin
- 一类受生物启发的双膦双硒镍配合物的合成及其电催化产氢性能
- Influence of NaOH Concentration on Sodium Storage Performance of Na0.44MnO2
- Photocathodic Protection on Stainless Steel by Heterostructured NiO/TiO2 Nanotube Array Film with Charge Storage Capability