
客体预嵌策略提升水系锌离子电池正极材料电化学性能
2021-04-02 02:23:42黄江涛周江梁叔全
物理化学学报 2021年3期
黄江涛,周江,2,*,梁叔全,2,*
1中南大学材料科学与工程学院,长沙 410083
2中南大学电子封装及先进功能材料湖南省重点实验室,长沙 410083

1 引言
可再生能源发电往往具有间歇性和随机性,未来电网需要大规模的储能系统对这种波动进行有效缓冲,以实现稳定的能量储存与输出1。在过去几十年里,锂离子电池作为高效、灵活的储能装置,在电子消费产品、电动汽车以及电网储能等领域得到了广泛的应用2,3。然而锂离子电池的生产成本较高,锂的资源储备不足,并且大多使用的是有毒、易燃的有机电解液,存在安全隐患的同时易造成环境污染,这些问题严重阻碍了其在大规模储能领域的发展和应用4。因此,人们开始寻求能够替代锂离子电池并兼具低成本、高能量、高安全性以及绿色环保等特点的新型储能电池体系。
与有机系电解液相比,水系电解液具有高离子电导率、高安全性、低成本、易封装等优势,近年来被广泛应用于Na+、K+以及多价金属离子如Mg2+、Al3+、Ca2+、Zn2+等二次电池体系中5–12。Na、K元素虽然储量丰富,成本低廉,但是其能量密度相对较低,离子半径较大,对正负极宿主材料的结构及性能要求更高,目前仍无法满足商业应用需求13–16。而多价金属离子能进行多电子转移反应,理论上可以提供更高的比容量。其中,金属锌因兼具高比容量(820 mAh·g-1)、低氧化还原电位(相对标准氢电极约为-0.763 V)、高析氢过电位、储量丰富以及无毒等优点而受到了广泛的关注17,18。自1988年Shoji等19报道了一种可充电Zn-MnO2水系电池以来,越来越多的专家学者开始对水系锌离子电池进行研究。区别于传统的碱性锌锰电池和镍锌电池,水系锌离子电池以中性或弱酸性的锌盐水溶液作为电解质(pH:3.6–6.0),对封装条件要求不高,生产成本低廉,安全且对环境友好,在大规模储能领域具有良好的发展前景20,21。
水系锌离子电池的储能机制主要基于负极金属锌的溶解与沉积22,23以及Zn2+、H+在正极宿主中的可逆嵌入与脱出24–26,部分正极材料通过溶解/沉积反应27,28或H+离子参与的化学转化反应进行储能29。近年来,关于水系锌离子电池正极材料的报道主要集中于钒、锰基化合物30–37、普鲁士蓝类似物38–40、有机化合物41–47以及部分过渡金属氧、硫化物等48–51。其中,具有层状、隧道状等晶体结构的钒、锰基化合物表现出了较强的锌离子存储能力52,53。钒和锰的多价态以及高氧化还原电位使其拥有相对较高的理论容量和放电电压平台(图1),应用前景十分广泛54。但与单价碱金属离子相比,二价锌离子具有更大的电荷密度,与正极宿主结构中阴离子的静电结合作用更强,扩散动力学相对缓慢55–57。例如,Li+在LiMn2O4等正极中的扩散系数为约为10-8–10-10cm2·s-1,而Zn2+在类似尖晶石结构中的扩散系数仅为10-11–10-13cm2·s-1,相差2至3个数量级58–60。同时,由于Zn2+难以从正极脱出,累积一定量后易引发正极的不可逆相变,导致初始结构的破坏61–63。正极结构组分在水系电解液中的溶解以及副反应产物的生成还会引起电池的容量衰减、循环稳定性较差64。此外,钒、锰基材料自身电导率低、倍率性能差等问题也严重限制了其发展35,65。

图1 不同水系锌离子电池正极材料相对锌负极的工作电压以及对应的比容量54Fig.1 Operating voltage versus specific capacity for Zn anode and various cathode materials presently used for aqueous ZIBs 54.
因此,为了获得高能量、高效率的水系锌离子电池以满足大规模储能应用需求,研究者对正极进行了深入的改性研究,并取得了一定的进展。其中在材料制备过程中,将金属阳离子、水分子等客体预嵌入正极的方法简便高效,在调控晶面间距、优化电子能带结构、提升反应动力学和循环稳定性等方面具有独特的优越性。本文总结了近年来利用客体预嵌策略提高钒、锰基正极材料性能的研究进展,分析了该策略的性能提升机制以及局限性,并对未来高性能水系锌离子电池钒、锰基正极材料的研究发展方向进行了展望。
2 钒基化合物
钒的资源储备丰富,具有+3、+4、+5多种价态和配位构型,其碳、氮、氧、硫化物及其衍生物被广泛地应用于高性能二次电池正极研究32,34,66–72。钒氧化物由V-O配位多面体组成,有层状(V2O5)或隧道状(VO2、V6O13)等多种晶体结构(图2a–c)64。早在上个世纪90年代,V2O5和V6O13的电化学嵌锌能力就已得到初步证实73–75。V2O5是由共用边角的VO5四方锥长链组成的单层结构,有利于锌离子的储存和迁移。然而随着锌离子的反复嵌入和脱出,这种不稳定的层状结构极易崩塌,导致循环稳定性较差76,因此V2O5作为水系锌离子电池正极的表现一直不尽如人意。直到2016年,Nazar等62报道了一种锌离子、水分子预嵌的双层σ-Zn0.25V2O5·nH2O纳米带,揭示了层间锌离子与结构水分子在充放电过程中的可逆交换机理,证明二者可以作为“支柱”显著提高V2O5的容量和循环稳定性之后,客体预嵌策略逐渐开始被广泛应用于水系锌离子电池正极材料的研究中。表1列出了近几年报道中的一些较为典型的客体预嵌钒基材料及其电化学性能,包括电压区间、电解液类型、Zn2+扩散速率、放电比容量以及循环性能。本节将对这些工作的研究重点进行介绍及讨论。

表1 一些典型的客体预嵌钒基材料的性能对比Table 1 Properties of some typical guest pre-intercalated vanadium-based materials.
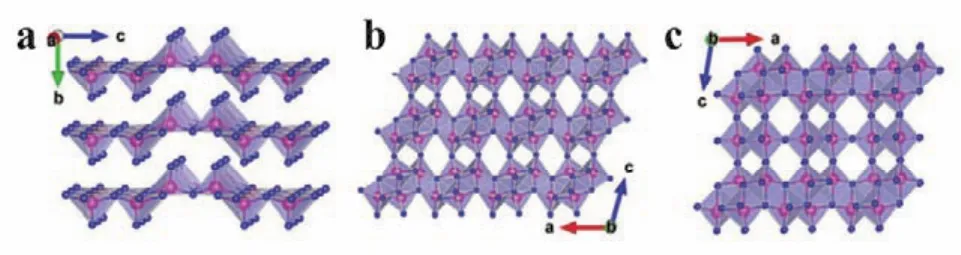
图2 (a) V2O5;(b) VO2;(c) V6O13的晶体结构64Fig.2 Crystal structures of (a) V2O5; (b) VO2; (c) V6O13 64.
2.1 水分子预嵌
在水系锌离子电池体系中,钒基材料的合成及应用环境大部分以水溶液为主,水分子对材料结构及性能的影响不容忽视。Yang和Mai等77通过溶胶凝胶法合成了一种V2O5·nH2O/石墨烯复合材料(VOG)作为水系锌离子电池正极,并系统地阐述了结构水分子在V2O5·nH2O中的作用。XRD结果显示,将VOG浸入Zn(CF3SO3)2电解液并充电至1.3 V后,晶面间距从1.26 nm缩小到了1.04 nm (图3a)。结合魔角旋转核磁共振谱中的1H、13C和19F谱峰变化可以得知(图3b),在充电状态下,层间嵌入的水分子、电解液离子(和Zn2+)与晶格氧之间形成了氢键,拉近了层间距离。当放电至0.2 V时,水合锌离子的嵌入使层间距扩大至1.35 nm,这与先前报道中Zn2+的嵌入使层间距缩小的结果完全相反62。实验结果表明,在这种机制的作用下,VOG的Zn2+扩散系数、能量密度以及倍率性能都要明显优于高温处理后不含结构水的VOG-350。同样,其他水分子预嵌的钒基氧化物,如层状结构的V5O12·6H2O、V3O7·H2O以及隧道状结构的V6O13等31,78–80,也表现出了优异的电化学储锌能力。结合密度泛函理论(DFT)计算可以得知,在充放电过程中,结构水分子的溶剂化作用“屏蔽”了Zn2+的部分电荷,使其与晶格氧之间的相互作用减弱,同时扩大了离子扩散通道,使Zn2+迁移能垒降低,反应动力学行为得到增强。

图3 (a) VOG在充电至1.3 V以及放电至0.2 V时的晶体结构示意图以及(b) MAS核磁共振谱77Fig.3 (a) Crystal structures and (b) MAS NMR spectra of VOG after charging to 1.3 V and discharging to 0.2 V 77.
2.2 阳离子预嵌
2.2.1 MxV2O5型
尽管水分子预嵌可以促进锌离子的扩散,但是充放电过程中V2O5的结构劣变仍会导致容量的迅速衰减,使得循环稳定性较差。继Zn2+预嵌的σ-Zn0.25V2O5·nH2O取得进展之后,众多金属阳离子预嵌的V2O5正极也相继被报道,如δ-Ca0.24V2O5·0.83H2O81和β-Na0.33V2O555等。通过金属离子的化学预嵌入,V2O5中的部分V5+被还原成V4+,结构稳定性和导电性得到显著提升。但是上述材料中的Zn2+、Ca2+和Na+已经改变了V2O5的本征结构,形成了新的V-O配位构型,如层状结构的σ型(4、5、6配位)、δ型(5、6配位)以及三维隧道结构的β型(5、6配位)。考虑到引入过量金属离子还会导致材料质量增大,比容量降低,本课题组探究了微量过渡金属离子(Fe2+、Co2+、Ni2+、Mn2+、Zn2+和Cu2+)的嵌入对V2O5结构稳定性、导电性以及离子扩散行为的影响82。例如,Cu0.1V2O5·0.08H2O的非原位XRD谱图表明,在充放电过程中,2θ= 7.7°处衍射峰对应的(001)晶面基本保持不变,说明Cu2+的引入增强了Zn2+嵌入/脱出时V2O5层状结构的稳定性,同时使部分V5+还原成V4+,提高了导电性和离子扩散能力,在20 A·g-1的大电流下循环3000次后仍可以保持122 mAh·g-1的容量。Li等83同样报道了一种微量Mn2+预嵌的Mn0.15V2O5·nH2O电极,其XRD谱图如图4a所示,并通过DFT计算分析了Mn2+对V2O5导电性的影响。结果表明,Mn2+能有效调控V2O5的电子能带结构,费米能级附近呈现出一个新的能级(图4b),使间接带隙从2.28 eV降至0.9 eV,导电性和倍率性能得到显著提升(图4c),在10和20 A·g-1下经过8000次的长循环后仍可以分别保持153和122 mAh·g-1的比容量。
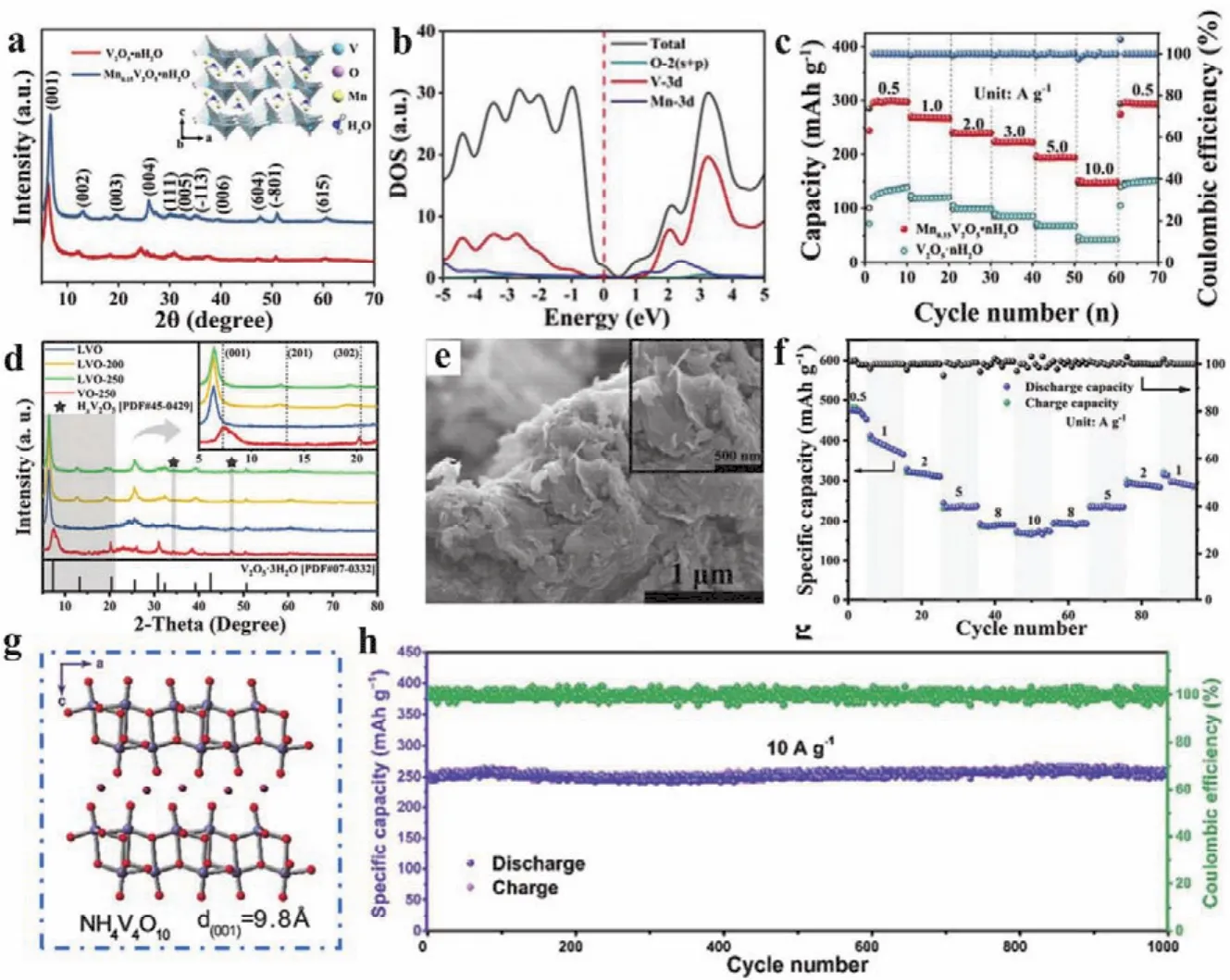
图4 Mn0.15V2O5·nH2O的(a) XRD图谱、(b)态密度图以及(c)倍率性能83;LVO-250的(d) XRD图谱,(e) SEM图像以及(f)倍率性能84;(g) NH4V4O10的原子结构以及(h)在10 A·g-1下的长循环性能63Fig.4 (a) The XRD pattern, (b) densities of states and (c) rate performance of Mn0.15V2O5·nH2O 83;(d) the XRD pattern, (e) SEM image and (f) rate capacities of LVO-250 84; (g) atom structure and(h) long-term cycling performance at 10 A·g-1 of NH4V4O10 63.
为了最大程度降低预嵌离子的摩尔质量对整体比容量的影响,本课题组还发展了Li+预嵌的LixV2O5·nH2O (LVO)正极材料(图4d)84。研究表明,在250 ℃空气煅烧过后的LVO-250具有不规则纳米层堆积的絮状形貌(图4e),结晶性较高。Li+的嵌入保持了V2O5·nH2O结构的完整性并使(001)晶面间距从1.20 nm扩大至1.38 nm,更有利于Zn2+的扩散。得益于Li+较小的摩尔质量以及LVO较大的层间距,其在0.5 A·g-1电流下的比容量高达470 mAh·g-1(图4f),并以10 A·g-1循环1000次后容量仍可达192 mAh·g-1。同理,将摩尔质量较小的NH4+作为结构支柱也取得了良好的效果:NH4V4O10表现出较大的晶面间距(0.98 nm) (图4g)和理想的锌离子扩散系数(1.79 × 10-9–1.27 × 10-8cm2·S-1),其中与V4O10层间形成的N-H···O氢键网络有效地提升了结构稳定性,因此可以在1 A·g-1的电流密度下贡献361.6 mAh·g-1的高比容量,并能以10 A·g-1稳定循环1000次以上(图4h)63。
除此之外,还有大量关于金属阳离子预嵌V2O5的研究报道,如Zn0.3V2O5·1.5H2O,MgxV2O5·nH2O和BaxV2O5·nH2O85,103–105等,但是对不同金属离子与V2O5之间形成的微观配位结构却少有研究,客体离子对锌离子储能行为的影响需要一个多尺度的表征和分析。Parkin和He等86对具有相同配位构型 的δ-Ni0.25V2O5·nH2O和δ-Co0.25V2O5·nH2O进 行了对比研究,并分别从原子和宏观电极结构的角度解释了两种材料不同电化学行为的原因。分析不同阶段的X射线吸收精细结构谱(NEAFX)后得知,在充放电过程中δ-Co0.25V2O5·nH2O中的Co有较为明显的不可逆脱出(图5a),非原位Raman和NEAFX测试结果也反映了键长和配位结构的改变,导致结构稳定性降低,循环性能较差。而δ-Ni0.25V2O5·nH2O在Zn2+的嵌入/脱出过程中则表现出高度可逆的相变行为以及优异的循环稳定性(图5b)。通过DFT计算表明,δ-Ni0.25V2O5·nH2O具有四种可能的Zn2+嵌入位点和两种扩散通道(图5c),锌离子的最高活化能垒仅为0.5 eV,并且利用可视化X射线断层扫描技术(CT)得到了δ-Ni0.25V2O5·nH2O电极的三维孔隙结构和扩散模拟图(图5d),结果表明该电极拥有较高的孔隙率和扩散系数,表现出了优于锂离子电池电极的反应传输能力。这也为今后对正极材料的储锌性能进行多尺度分析开拓了新视野。

图5 (a) Co0.25V2O5·nH2O中Co以及(b) Ni0.25V2O5·nH2O中Ni在不同状态下的L边X射线吸收精细结构谱图;(c) Ni0.25V2O5·nH2O中Zn2+的4种嵌入位点和2种扩散通道以及(d)基于电极的孔结构进行扩散模拟得到的通量分布图86Fig.5 L-edge NEXAFS spectra at different stages of (a) Co in Co0.25V2O5·nH2O and (b) Ni in Ni0.25V2O5·nH2O;(c) four intercalation sites and two diffusion channels for Zn2+ ions and (d) the flux distribution in diffusion simulation based on the pore morphology of Ni0.25V2O5·nH2O electrode 86.
在已有的嵌入型机制的基础上,探索新型的储能机制也是获得高能量、高功率密度水系锌离子电池的有效途径。为此,本课题组首次报道了一种基于Zn/Ag0.4V2O5电池的置换/嵌入相结合的储锌机制87:在放电初期,部分Ag+被Zn2+置换,生成新相Zn2(V3O8)2和金属相Ag0;随着放电过程的进行,Zn2+分别嵌入Zn2(V3O8)2和Ag0.4V2O5两相的位点中。其中新相Zn2(V3O8)2为Zn2+提供了更多的结合位点,而Ag0的原位生成为整个电极提供了良好的导电网络。这种新型的储锌机制使Ag0.4V2O5在20 A·g-1的大电流下稳定循环4000次后,仍可以保持144 mAh·g-1的比容量。
然而钒基材料的工作电压普遍较低(~0.7 V),近80%的容量贡献都来源于1.0 V以下的电化学反应,虽然比容量较高,但能量密度(低于250 Wh·kg-1)远达不到应用要求。Zhi等88通过钴离子的嵌入以及“盐包水”型电解液的使用,成功地将V2O5的输出电压提升至1.7 V (图6a),其中高于1 V的容量达到227 mAh·g-1,占总容量的52.54%。理论计算显示,Co0.247V2O5·0.944H2O中Co 3d和V 3d轨道间的相互作用提高了V5+/V4+的氧化还原电位(图6b),且由于Co 3d、V 3d和O 2p轨道间存在pd杂化,Zn2+的有效电荷得到降低(2→1.35),其Zn2+吸收能从1.85 eV增加到了2.24 eV,表现出优异的Zn2+扩散能力(图6c,d)。使用20 mol·L-1LiTFSI +1 mol·L-1Zn(CF3SO3)2型电解液时,水的分解得到了充分抑制,因此实现了电池的高工作电压和优异的倍率性能,在10 A·g-1的电流密度下能贡献163 mAh·g-1的比容量,并且循环7500次后容量保持率仍高达90.26%。

图6 (a) Co0.247V2O5·0.944H2O与V2O5·nH2O的恒流充放电曲线以及(b)能量和态密度的对比图;(c) Co0.247V2O5·0.944H2O和(d) V2O5·nH2O的Zn2+吸收能对比88Fig.6 (a) Galvanostatic charge/discharge curves and (b) the energy versus DOS of Co0.247V2O5·0.944H2O and V2O5·nH2O cathodes; the adsorption energy of Zn2+ for (c) Co0.247V2O5·0.944H2O and (d) V2O5·nH2O 88.
2.2.2 MxV3O8型
在层状MxV3O8型钒酸盐结构中,V3O8层由共角的VO6八面体和VO5四方锥组成,层与层之间由金属离子M (Li+、Na+、K+等)连接并作为稳定结构的支柱。Kim等89研究了LiV3O8在Zn2+嵌入及脱嵌时经历的相转变过程。整个放电过程分为两个阶段,在放电初期,Zn2+逐渐占据Li(2)位点形成ZnLiV3O8相,而在放电后期占据Li(3)位点形成ZnyLiV3O8(y> 1)相,充电过程则随Zn2+的脱出完成从ZnyLiV3O8到LiV3O8的逆向转变。这种储锌机制使LiV3O8能在133 mA·g-1的电流密度下贡献172 mAh·g-1的容量。
钒酸钠的常见构型除了上节中提到的隧道状β-Na0.33V2O5型外,还有层状的NaV3O8型,在这两种结构中Na+都能作为有效“支柱”来保持结构稳定性。为了探究不同组分和构型对Zn2+储存机理的影响,本课题组构建了两种NaV3O8型层状结构 (Na5V12O32和 HNaV6O16·4H2O) 和 一 种β-Na0.33V2O5型隧道结构(Na0.76V6O15)的钒酸钠(图7a,b)90。对比循环曲线(图7c)和循环伏安曲线(图7d–f)得知,层状型结构更有利于Zn2+的扩散,容量较高,但循环稳定性差;隧道型结构的循环稳定性较高,但容量偏低。这是因为层状结构中的氧原子分为单键连接和三键连接,而隧道结构中的氧原子仅为单键连接,与Na+的结合作用更强,结构更稳定。并且由于Zn2+的半径比Na+小,具有更高的电荷密度,容易取代层状Na5V12O32中与三键氧原子结合的Na+,形成新相Zn4V2O9。而被取代的Na+则扩散至电解液中,造成层状结构的崩塌和容量的衰减。
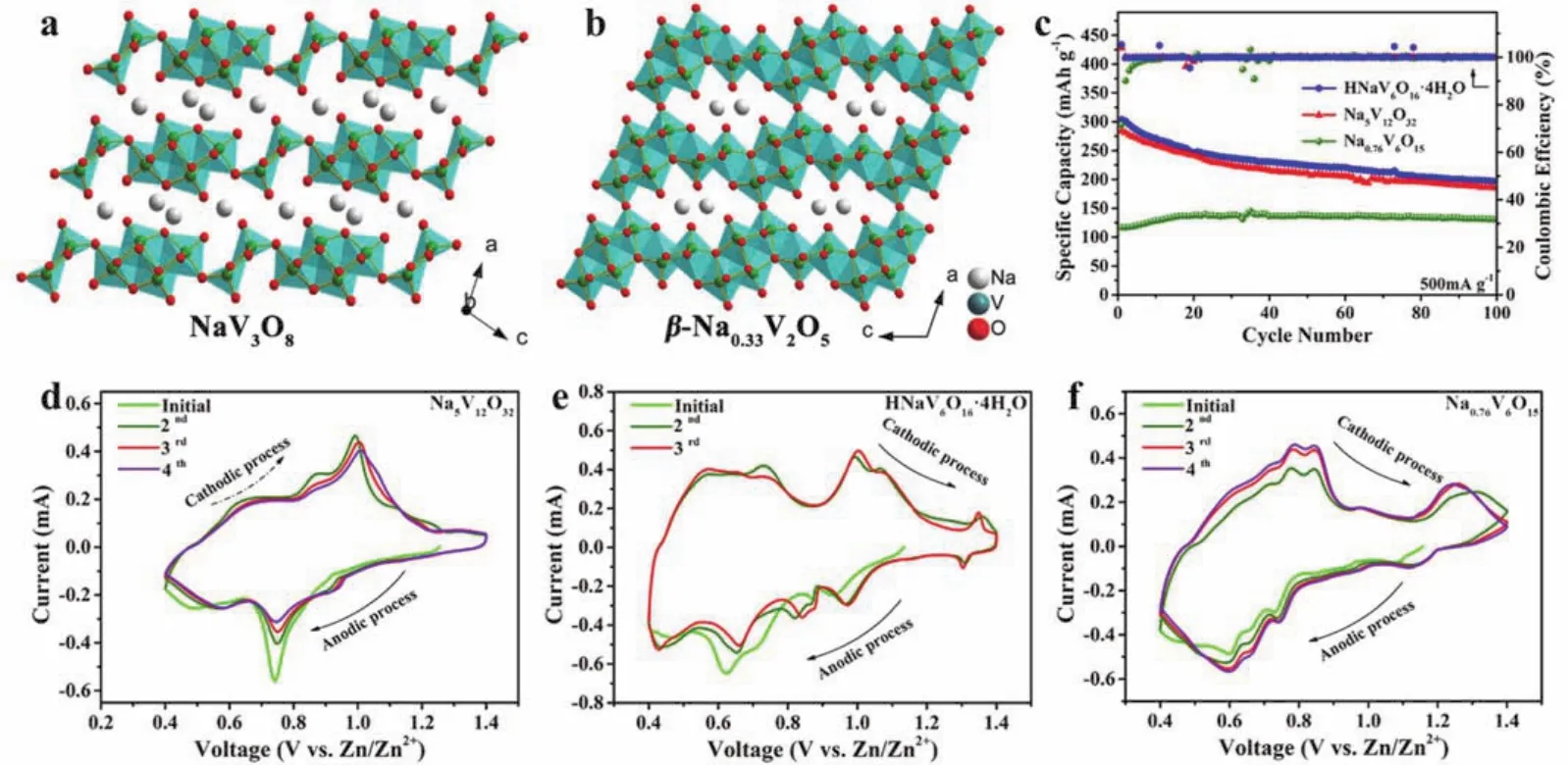
图 7 (a) NaV3O8和(b) β-Na0.33V2O5的晶体结构;(c) Na5V12O32,HNaV6O16·4H2O 和 Na0.76V6O15在500 mA·g-1电流下的循环性能以及(d–f)循环伏安曲线90Fig.7 The crystal structure of (a) NaV3O8 and (b) β-Na0.33V2O5; (c) cycling performances at 500 mA·g-1 and CV curves of (d) Na5V12O32, (e) HNaV6O16·4H2O and (f) Na0.76V6O15 90.
将 一系列层状结构(KV3O8和K2V6O16·1.57H2O)和隧道结构(K2V8O21和K0.25V2O5)的钒酸钾应用于锌离子电池中也得到了相似结论:在K2V8O21和K0.25V2O5中,K+发挥着稳定隧道结构、提高锌离子储存能力的重要作用,其中K2V8O21的容量最高,K0.25V2O5的循环性能最好;而层状的KV3O8和K2V6O16·1.57H2O则更容易在Zn2+嵌入/脱出过程中遭受结构破坏,其快速的容量衰减以及较差的循环伏安曲线重合度证明了这一点91。
因此,为了提高层状钒酸盐的结构稳定性,减少活性物质在电解液中的溶解,Niu等92采用Na2SO4作为电解液添加剂,显著抑制了NaV3O8·1.5H2O中Na+的脱出和钒的溶解(图8a),同时还实现了Zn2+和H+在NaV3O8·1.5H2O中的同步嵌入和脱嵌,使其能在1 A·g-1的电流下输出221 mAh·g-1的比容量并稳定循环100次(图8b),且容量保持率高达90%。而在相同测试条件下,采用只含ZnSO4的电解液时容量保持率仅为34%,表现出较差的循环稳定性。
与单价碱金属离子相比,多价金属离子与晶格氧之间的键合作用更强,对结构稳定性会有更明显的提升。Huang等93首次报道了半径相近的Na+和Ca2+(约为0.1 nm)选择性共嵌入的V3O8型结构(NaCa0.6V6O16·3H2O)。在钠、钙离子和结构水分子的协同作用下,Zn2+沿b轴的迁移能垒成功降低至0.89 eV (图8c,d),表面电荷转移以及Zn2+在电极内部的扩散能力都得到了显著提升,表现出优异的倍率性能。吉布斯自由能计算结果表明,在充放电过程中,Na+和Ca2+不易被Zn2+取代,具有较高的稳定性。该材料以2 A·g-1充放电时最高容量为231 mAh·g-1,并且经过2000次循环后仍具有94%的容量保持率。
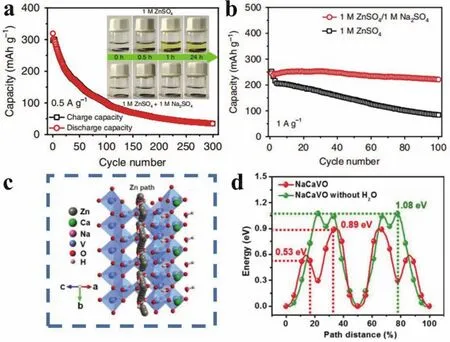
图8 (a) NaV3O8·1.5H2O在ZnSO4电解液中的循环性能(0.5 A·g-1)以及(内置)在不同电解液中静置后电解液的颜色变化;(b) 1 A·g-1下 NaV3O8·1.5H2O 在两种电解液中的循环性能 92;Zn2+在 NaCa0.6V6O16·3H2O 中的(c)扩散路径以及(d)迁移能垒93Fig.8 (a) Cycling performance of NaV3O8·1.5H2O in ZnSO4 at 0.5 A·g-1 and (inset) optical images of electrolytes containing NaV3O8·1.5H2O for different periods; (b) cycling performance of NaV3O8·1.5H2O at 1 A·g-1 in two electrolytes 92; (c) potential path and (d) energy barrier of Zn2+ diffusion in NaCa0.6V6O16·3H2O 93.
钒酸银因具有钒和银两种金属活性中心,在二次电池储能领域展现出了较大的应用前景。如上节内容所述,在放电过程中,Ag+可被Zn2+取代,生成具有高导电性的金属Ag0,而V5+则随着Zn2+的嵌入被部分还原成V4+。然而对于不同晶体结构、不同Ag/V比的钒酸银来说,其电化学行为也有着明显区别:层状结构的Ag1.2V3O8表现为Zn2+的可逆嵌入/脱出反应和Ag+的不可逆置换反应,在完全充电状态能检测到金属Ag0相的存在,具有较高的容量和倍率性能;隧道结构的Ag0.33V2O5表现为可逆的嵌入/脱出和置换反应,循环稳定性较好;链状的β-AgVO3因无法承受放电过程中的应力变化,最终转变成含Ag0的无定型相,表现为不可逆的嵌入/脱出和置换反应106。这些现象为从晶体结构层面探索水系锌离子电池的新型储能行为提供了新视角。
2.2.3 MxV2O7型
在钒基材料中,Zn2+与晶格之间较强的静电结合作用通常会诱导不可逆相变的发生,致使一些新相如Zn3V2O7(OH)2·2H2O的生成63。Zn3V2O7(OH)2·2H2O是由ZnO6八面体层与层间支柱组成的开放式框架结构(图9a)。当其作为副反应产物出现时,由于Zn2+难以再次从ZnO6八面体中脱出,会导致电极的电化学活性降低,容量衰减迅速。但将其直接作为Zn2+宿主材料时,这种稳定的层状结构反而有利于额外Zn2+的存储与扩散。基于此,Alshareef等94报道了一种超长的Zn3V2O7(OH)2·2H2O纳米线,随着Zn2+的逐步嵌入最终得到放电产物Zn4.9V2O7(OH)2·2H2O,并且在50和3000 mA·g-1的电流下可以分别贡献213和76 mAh·g-1的比容量(图9b)。Kim等107也报道了一种具有相似层状结构的α-Zn2V2O7纳米线,能量密度达到166 Wh·Kg-1,以4 A·g-1的电流密度循环1000次以后仍具有85%的容量。
与Zn3V2O7(OH)2·2H2O的嵌入/脱出机制不同,Cu3V2O7(OH)2·2H2O还表现出了在钒酸银中常见的还原置换反应机制(图9c):Zn2+的嵌入促使Cu2+还原成金属Cu0颗粒,同时生成新相Zn0.25V2O5·H2O,当Zn2+脱出时则恢复至Cu3V2O7(OH)2·2H2O结构,仅有少量的Cu0颗粒保留108。同样,在CuV2O6结构中也能观察到类似的置换机制:当Zn2+取代VO6八面体间的Cu2+后,形成具有相同结构的ZnV2O6和均匀分布的Cu0。这种置换方式既保证了晶体结构的总体稳定性,又通过原位生成的Cu0形成了导电网络,有效地提升了整个电极的循环稳定性和倍率性能95。

图9 Zn3V2O7(OH)2·2H2O的(a)晶体结构以及(b)倍率性能94;(c) Cu3V2O7(OH)2·2H2O的还原置换反应机制108Fig.9 (a) Crystal structure and (b) rate performance of Zn3V2O7(OH)2·2H2O 94; reduction displacement reaction mechanism of Cu3V2O7(OH)2·2H2O 108.
2.2.4 其他MxVmOn型
此外,还有许多不同构型的阳离子预嵌钒氧化物被相继报道。Al3+的预嵌使层状H11Al2V6O23.2拥有较大的(001)晶面间距(1.34 nm),有利于Zn2+的扩散,与石墨烯复合后进一步提高了电极的导电性和结构稳定性96。尖晶石结构的ZnV2O4表现出独特的电化学活化机制:在初始循环过程中,ZnV2O4经历了从体相到微晶相的转变,界面反应动力学得到增强,表现出优异的倍率性能和长循环稳定性109。层状结构的Fe5V15O39(OH)9·9H2O具有Fe和V两种电化学活性中心,在充放电过程中进行Fe3+/Fe2+、V5+/V4+以及V4+/V3+的电子转移反应,因此可以在0.1 A·g-1的电流密度下提供385 mAh·g-1的高比容量110。这些基于不同预嵌阳离子、晶体结构以及储能机理的研究推动了钒基材料在水系锌离子电池领域的迅速发展。
2.3 阴离子预嵌
近年来,应用于水系锌离子电池的钒基材料大都基于钒离子的氧化还原反应,平均工作电压普遍低于1.0 V,能量密度较低,仍无法达到实际应用需求。为了实现高工作电压、高能量密度的水系锌离子电池,引入阴离子氧化还原机制是一种可能的策略。要想解锁钒氧化物中的氧使其参与反应,需要对V-O共价键进行弱化,使O的电荷密度增加,电化学活性得到增强。考虑到引入键能更低的P-O共价键可以提升VxOy层中O的相对电荷密度,Chen和Niu等97设计了一种采用“盐包水”型电解液的Zn/VOPO4电池,解锁了O2-/O-在高电压区间的氧化还原反应,成功将平均工作电压提升至1.56 V (图10a)。本课题组也开发出一种氮氧化钒(VNxOy),实现了OH-在VNxOy表面的吸附/释放以及N3-/N2-的氧化还原反应(图10b,c),在30 A·g-1的大电流下可以释放200 mAh·g-1的比容量,并能以20 A·g-1稳定循环2000次以上69。岩盐氮氧化钒(VN0.9O0.15)具有紧凑的面心立方结构,原本不利于锌离子的扩散,但经过首次充电的电化学活化后,部分高价态的N3-会被低价态的O2-取代,从而形成具有丰富空位和缺陷的阴离子无序岩盐结构(VN0.2O2.1),极大地促进了锌离子的扩散(图10d),因此可以在0.2C的电流密度下输出603 mAh·g-1的超高比容量,并表现出优异的倍率性能111。

图10 (a) Zn/VOPO4电池在0.05 A·g-1下的充放电曲线97;(b) VNxOy表面的漫反射傅立叶变换红外光谱图以及(c)不同状态下N的近K边X射线吸收精细结构光谱69;(d) VN0.2O2.1的Zn2+扩散系数111Fig.10 (a) Charge/discharge curves at 0.05 A·g-1 of Zn/VOPO4 batteries 97; (b) DRIFT spectra of electrode surface and(c) N K-edge NEXAFS spectra of VNxOy at different states 69; (d) Zn2+ diffusion coefficient of VN0.2O2.1 111.
NASICON型结构的钒磷酸盐具有开放的三维框架,其间隙孔道有利于Li+、Na+的扩散,因此在锂、钠离子电池中得到了广泛的应用112。Huang等98将Na3V2(PO4)3应用于水系锌离子电池正极,通过首次充电将18e位点的Na+脱出后,得到具有Zn2+嵌入位点的NaV2(PO4)3,从而实现了从NaV2(PO4)3到ZnxNaV2(PO4)3的可逆相转变。在0.5C下,Na3V2(PO4)3/C复合材料可以在接近1.1 V的放电平台贡献97 mAh·g-1的比容量,100次循环后仍具有74%的保持率。Jiang等99将具有强电负性的F-引入NASICON型结构中,使Na3V2(PO4)2F3的工作电压成功提升至1.62 V,采用2 mol·L-1Zn(CF3SO3)2电解液时,能量密度可达97.5 Wh·kg-1,循环4000次后容量衰减仅为4%。尽管NASICON型结构具有较高的工作电压,但由于阴离子数量较多,材料摩尔质量较大,导致比容量较低,目前仍无法满足实际应用需求。
2.4 有机分子预嵌
除阴、阳离子外,中性的有机分子也可以作为客体扩大钒基宿主材料的层间距,提高其晶体结构稳定性。例如,Xia等100报道了一种聚3,4-乙烯二氧噻吩(PEDOT)预嵌的钒酸铵,使NH4V3O8的层间距从初始的0.78 nm扩增至1.08 nm。其中,层间的PEDOT既能作为结构支柱,又能提供良好的导电网络,同时还引入了氧缺陷,极大地提升了NH4V3O8的容量和循环稳定性。Li等113也报道了一种共轭导电聚合物聚苯胺(PANI)预嵌的V2O5,成功将(001)晶面扩大至1.40 nm,促进了Zn2+的扩散,并且苯胺分子的引入使V2O5中的部分V5+被还原成了V4+,本征导电性得到提高。值得注意的是,在首次放电的初期阶段,锌离子的嵌入会诱导副反应产物Zn3(OH)2(V2O7)·2H2O的生成,但在放电完成阶段却检测不到该相的存在,说明该产物只能在某一特定的Zn2+浓度范围内保持热力学稳定。然而目前还没有相关报道能对该现象提出合理的解释,未来还需要更进一步的研究。
如前几节内容所述,磷酸根离子以及水分子的预嵌可使层状结构的VOPO4·2H2O获得较高的Zn2+嵌入电位和较宽的晶面间距。但在锌离子嵌入/脱出过程中,其结构变化严重,容量衰减较快。为此,Srinivasan等101采用聚吡咯(PPy)预嵌的方法,显著提高了VOPO4·2H2O的循环性能和倍率性能。与其他有机分子预嵌使层间距扩大的结果不同,PPy-VOPO4(0.67 nm)表现出了比VOPO4·2H2O更小的晶面间距(0.74 nm)。这是因为与水分子相比,PPy分子与VOPO4层间的结合作用更强,形成的结构更稳定。由此可知,晶面间距不一定与材料的电化学性能成线性关系,在保证正极宿主结构稳定的前提下,一定程度的晶面间距缩小并不会影响Zn2+的扩散能力。
Gu等102将十二烷胺分子引入V-O层中,得到了C12-VOx纳米管。与传统的层间距调控机制不同,经过放电初期的原位活化后,C12-VOx逐渐转变为层状结构的Zn3V2O7(OH)2⋅2H2O和非晶相组织Znn-VOx-C12,并且随着Zn2+的进一步嵌入,最终完全转化为非晶相的Znn-VOx-C12。而在充电过程中,随着Zn2+的脱出,层状结构的Zn3V2O7(OH)2⋅2H2O能够得以恢复。在这种转化/嵌入机制下,C12-VOx能够以2.4 A·g-1的电流密度稳定循环近1000次,并且具有242.5 Wh·kg-1的高能量密度。
3 锰基化合物
锰在自然界中的储备十分丰富,且成本低廉、毒性低、对环境友好,具有多种氧化态(Mn2+,Mn3+,Mn4+和Mn7+),在储能领域表现出了广阔的应用前景。以MnO2为首的众多锰基氧化物(如Mn3O4,Mn2O3和MnO等)以及锰酸盐(如ZnMn2O4等),都被证实了具有一定的Zn2+储存能力25,53,114–119。MnO2的晶体结构取决于MnO6八面体的连接方式,包括α,β,γ,δ和λ型等(图11)64。其中具有隧道结构的α-MnO2(2 × 2, 0.46 nm × 0.46 nm),γ-MnO2(1 ×1,0.23 nm × 0.23 nm和1 × 2,0.23 nm × 0.46 nm)以及层状结构的δ-MnO2(0.7 nm)比较有利于Zn2+的嵌入与扩散。MnO2拥有较高的理论比容量(Mn4+/Mn2+,616 mAh·g-1)和高于钒基材料的工作电压(约为1.3 V),应用前景较好。然而大部分被报道的以MnO2为主的锰基材料,它们的实际容量都要远低于理论容量,并且储能机理复杂,衍生副反应较多,例如,α-MnO2随着Zn2+和H+的共嵌入会发生向ZnxMnO4、MnOOH和Mn2O3的转变,体积变化严重120;充放电过程中Mn3+的John-Teller效应还会诱发晶格畸变和歧化反应,导致二价Mn2+的溶解和不可逆相的生成,严重影响了锰基材料的循环稳定性29,121,122。同时,锰基材料的电导率较低,倍率性能通常不够理想。为了解决这些问题,研究人员同样采用客体离子或分子预嵌的策略,显著提高了锰基材料作为水系锌离子电池正极的电化学性能。不同客体预嵌不同晶型锰基材料的性能如表2所示。

表2 不同客体预嵌锰基材料的性能对比Table 2 Properties of some typical guest pre-intercalated Mn-based materials.
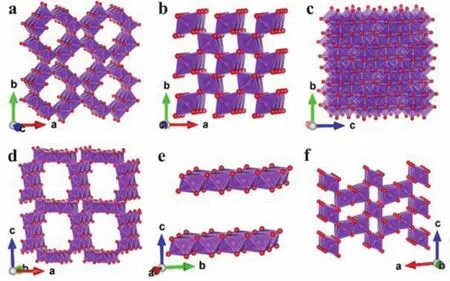
图 11 (a) α-MnO2;(b) β-MnO2;(c) λ-MnO2;(d) todorokite MnO2;(e) δ-MnO2;(f) γ-MnO2的晶体结构 64Fig.11 Crystal structures of (a) α-MnO2; (b) β-MnO2;(c) λ-MnO2; (d) todorokite MnO2; (e) δ-MnO2; (f) γ-MnO2 64.
3.1 水分子预嵌
水钠锰矿型二氧化锰(δ-MnO2)的层间距较大,有利于Zn2+的嵌入与扩散,其容量通常比隧道型的二氧化锰更高,但在循环过程中容易发生结构劣变,导致新相的不可逆生成(如MnO、MnOOH和ZnMn2O4等)和容量的迅速衰减,并且Zn2+与晶格氧之间的强静电作用会造成Zn2+的扩散动力学缓慢,严重限制了其在水系锌离子电池中的发展。因此,Choi等123通过电化学转化法合成了一种高层间水分子含量(质量分数约为10%)的δ-MnO2,极大地提升了层状结构的稳定性,并抑制了其循环过程中向尖晶石结构的转变,表现出优异循环性能和倍率性能,在100 mA·g-1的电流密度下可以获得350 mAh·g-1的高比容量。理论计算显示,在水分子的配位作用下,Zn2+在层间的迁移能垒仅为0.25 eV (图12a,b),远低于尖晶石结构中未水化Zn2+的迁移能(1.03 eV),并且由于层间距较小(0.7 nm),脱离出的Mn2+无法形成可自由移动的[Mn(H2O)6]2+,而是以一种稳定的Zn-Mn“哑铃”构型存在于层间(图12c),从而抑制了Mn的溶解。由此可知,高水分子含量以及适当的晶面间距可以协同提升δ-MnO2的循环性能,这对于其他晶体结构的正极材料同样具有启示意义。
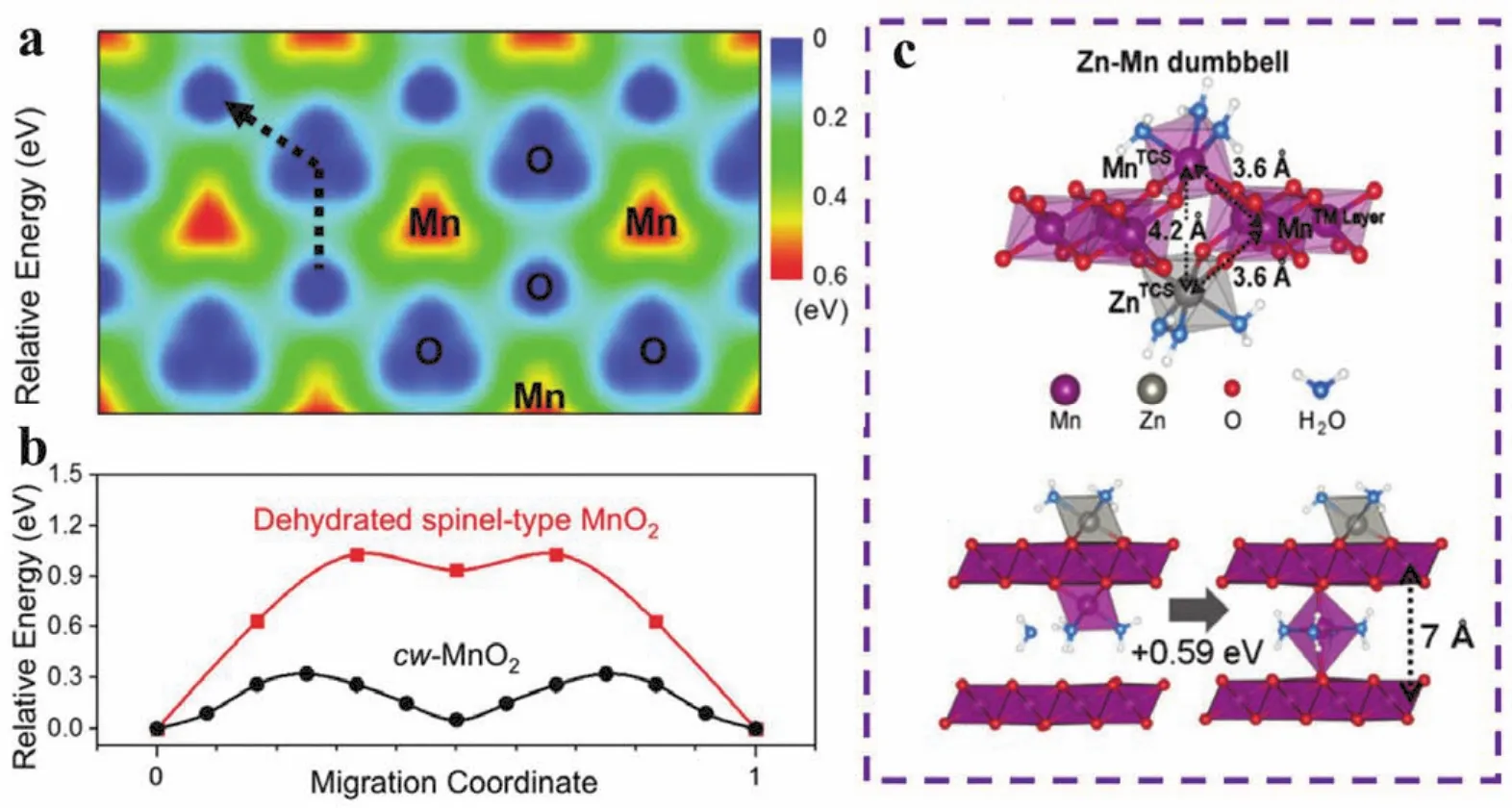
图12 水合Zn2+在δ-MnO2层间的(a)势能面以及(b)迁移能垒;(c) Zn-Mn“哑铃”构型的示意图以及Mn八面体在层间距为0.7 nm时的配位倾向123Fig.12 (a) The potential energy surface and (b) migration barrier with respect to hydrated Zn2+ in the interlayer space of δ-MnO2; (c) the energetically complexation behavior of Mn octahedral with interlayer distance of 0.7 nm 123.
3.2 阳离子预嵌
3.2.1 隧道结构
最近的研究表明,在电解液中添加额外的Mn2+可以抑制Mn3+的歧化反应,从而提升锰基材料的循环稳定性24。通过表面石墨烯包覆的手段也可以有效缓解体积膨胀,降低锰的溶解率76,124。然而,如何从锰基材料的本征结构出发来改善其结构稳定性,仍需要进一步的研究。
本课题组通过研究发现,将K+预嵌入α-MnO2的(2 × 2)隧道中形成K0.8Mn8O16可以有效抑制循环过程中Mn的溶解125,在50次循环内,电解液中Mn2+含量相比纯相α-MnO2更稳定(图13b)。这是由于隧道中的K+与晶格氧之间形成了较强的键合作用,结构稳定性得到增强(图13a)。此前有报道称,MnO2隧道内较大的阳离子(如K+、Ba2+等)会形成物理阻挡和静电排斥力,阻碍金属离子的扩散126。而在K0.8Mn8O16中,氧缺陷的存在打开了MnO6多面体墙,为H+的扩散提供了额外通道(以H+嵌入机制为主),极大地提升了电化学活性和反应动力学(图13c)。因此在K+和氧缺陷的协同作用下,K0.8Mn8O16展现出了398 Wh·kg-1的高能量密度和超过1000次的循环稳定性。
从理论上来说,进一步提高α-MnO2中K+的含量可以缓解因K+从隧道中脱出而引起的结构不稳定性。Zhang等127通过研究发现,将高K+含量的α-K0.19MnO2作为水系锌离子电池正极时,其表现为H+/Zn2+共嵌入机制(图13d),并随着H+和Zn2+的逐步嵌入,α-K0.19MnO2经历了从隧道结构到层状结构的相转变,且层间的K+作为结构“支柱”有效地提升了结构稳定性,促进了H+和Zn2+的扩散,因此可以在20C的大电流密度下输出113 mAh·g-1的比容量(图13e)。
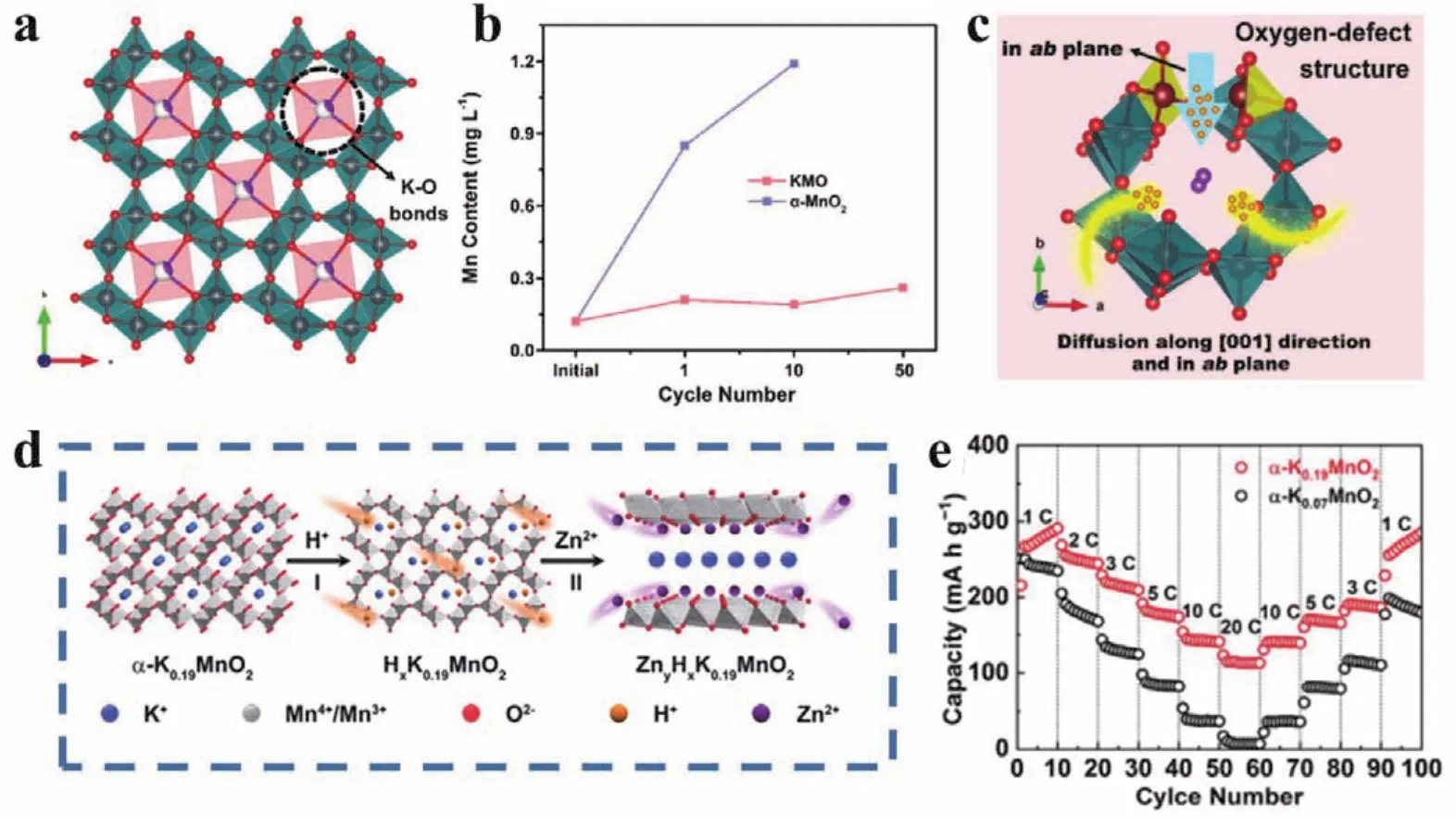
图13 (a) K0.8Mn8O16的晶体结构图;(b) K0.8Mn8O16和α-MnO2在循环过程中电解液所含Mn2+的浓度变化125;(c) H+在K0.8Mn8O16氧缺陷结构中的扩散示意图;α-K0.19MnO2的(d)储能机理以及(e)倍率性能127Fig.13 (a) Crystal structure of K0.8Mn8O16; (b) concentration of Mn2+ in electrolyte during cycling of K0.8Mn8O16 and α-MnO2; (c) schematic illustration of H+ diffusion in structure with oxygen defects 125; (d) energy storage mechanism and (e) rate performance of α-K0.19MnO2 127.
然而α-MnO2从隧道结构到层状结构的转变会造成严重的体积变化,这对于锌离子电池的循环寿命来说是非常不利的。为了实现更加稳定的储能,Mai等128将Zn2+预嵌的α-MnO2(ZnxMnO2)与柔性碳布进行复合,制备了一种具有电池级能量密度的锌离子混合超级电容器,并获得了优异的电化学性能。
Todorokite型MnO2具有3 × 3大尺寸隧道结构,通常需要金属离子或水分子来保持结构稳定。Lee等129报道了一种Mg2+和水分子共嵌的Todorokite-MnO2(Mg1.8Mn6O12·4.8H2O)作为水系锌离子电池正极,在0.5C的电流密度下可输出108 mAh·g-1的比容量,并且具有良好的循环稳定性和倍率性能。这是由于隧道内含有大量的Mg2+和水分子作为结构支柱,阻止了向其他结构的转变。同时,水分子的“屏蔽”作用降低了Zn2+的有效电荷,促进了Zn2+的扩散。但由于其容量较低,储能机理仍不明确,未来仍需进一步研究。
3.2.2 层状结构
为了提高δ-MnO2的储锌性能,Wu等130通过固相法合成了一种Na+预嵌的层状Na0.95MnO2结构,使用0.5 mol·L-1Zn(CH3COO)2和 0.5 mol·L-1CH3COONa作为电解液时具有1.4 V的平均电压平台,并且能够在1–2 V的电压窗口以4C的电流密度稳定循环1000次。同样,受阳离子预嵌层状钒基材料的启发,Yu等122采用氢氧化钠溶液选择性刻蚀硅酸锰的方法,将Na+、水分子和MnO6八面体进行重排,得到Na+和水分子共嵌入的层状锰酸钠Na0.55Mn2O4·0.57H2O (NMOH)。其中,Na+和水分子作为支柱扩大了MnO6层的间距,提高了结构稳定性以及Zn2+的扩散能力。NMOH表现为Zn2+和H+的置换/嵌入的储能机制:在首次循环中,部分Zn2+将取代Na+作为层间支柱,促进后续循环中Zn2+和H+的可逆嵌入与脱出;而被置换出的Na+则通过NMOH的表面吸附作用,以赝电容的形式贡献少部分容量(图14a)。这种独特的储能机制使NMOH能在200 mA·g-1的电流密度下贡献389.8 mAh·g-1的比容量,并且在200至1500 mA·g-1的电流范围内表现出了优异的倍率性能(图14b)。该实验结果进一步证明了,采用金属离子和水分子共嵌的策略可以有效地提升锰基材料作为水系锌离子电池正极的电化学性能。

图14 (a) Na0.55Mn2O4·0.57H2O的储锌机制示意图以及(b)其在200到1500 mA·g-1电流范围内的倍率性能122Fig.14 (a) Schematic illustration of the zinc storage mechanism of Na0.55Mn2O4·0.57H2O and(b) its rate performances from 200 to 1500 mA·g-1 122.
在钒基材料中,Zn2+、Ca2+等二价金属离子作为支柱时可以更有效地提高层状结构的稳定性,但很少有研究将其应用于锰基材料中。基于该理念,Wang等131构建了一种Zn2+和水分子预嵌的层状MnO2纳米花。这种结构的MnO2具有高比表面积和高介孔率,活性位点较为丰富,并且在Zn2+和水分子的协同作用下,结构稳定性和离子扩散系数得到显著提升,因此表现出良好的循环性能和倍率性能。Tao等132报道了一种Ca2+和水分子预嵌的δ-MnO2(Ca0.28MnO2·0.5H2O),表现为H+和Zn2+共嵌入/脱出的储能机理。在Ca2+与水分子的协同作用下,该正极能在3.5 A·g-1的电流密度下循环5000次后几乎没有容量衰减,突显了客体预嵌策略的优越性。
3.2.3 尖晶石结构
对于具有完整尖晶石结构的锰酸盐(MxMn2O4)而言,金属离子的预嵌使其本身具有较高的结构稳定性,但由于晶格中的Mn3+对Zn2+的静电斥力较大,Zn2+的迁移能垒较高,通常不适宜作为Zn2+的宿主材料。Chen团队59通过一种温和的液相合成法,成功将阳离子缺陷引入尖晶石ZnMn2O4中,促进了Zn2+在这种缺陷结构中的扩散动力学行为(图15a,b)。丰富的Mn空位不仅使尖晶石结构的Zn2+扩散系数提升了2个数量级(图15c),同时显著降低了电荷转移阻抗(从1000 Ω降至280 Ω),导电性得到极大提升。使用3 mol·L-1Zn(CF3SO3)2电解液时,大体积的离子和较高的盐浓度降低了水的活性,有效地抑制了Mn的溶解。XRD测试结果表明,在充放电过程中,这种缺陷结构十分稳定,仅表现为晶格的收缩与膨胀,并具有优异的循环性能。
其他阳离子预嵌的尖晶石结构也展现了一定的储锌潜力。例如,Kim等136基于MgMn2O4正极(图15d)开发了一种高电压、高能量的混合双离子电池,实现了Mg2+和Zn2+的共嵌入。首次充电后,预嵌的Mg2+被抽离出尖晶石结构,为后续循环中Mg2+和Zn2+的嵌入提供了活性位点。使用包含ZnSO4,MgSO4和MnSO4的混合电解液时,平均工作电压达到1.5 V (图15e),以500 mA·g-1的电流密度循环时表现出了良好的循环稳定性,且经过500次循环后仍保持96 mAh·g-1的比容量(图16f)。值得一提的是,在完全充电和放电阶段分别观察到了(MgMn)9Zn4(OH)22(SO4)2·8H2O和Zn4(OH)6SO4·0.5H2O两种副产物的生成,但它们在整个电化学反应过程中所扮演的角色仍不明确,因此需要进行更深入的探讨。
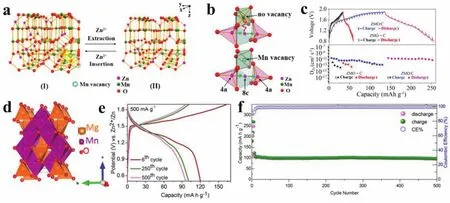
图15 (a) Zn2+在ZnMn2O4尖晶石结构中的嵌入/脱出示意图;(b) Zn2+在完整以及含Mn空位的ZnMn2O4结构中的扩散路径59;(c) ZnMn2O4/C与ZnMn2O4 + C的GITT曲线以及计算得到的Zn2+扩散系数;(d) MgMn2O4的晶体结构图以及在500 mA·g-1电流密度下(e)不同圈数的充放电曲线和(f)相应的循环性能图136Fig.15 (a) Schematic illustration of Zn2+ insertion/extraction in ZnMn2O4 spinel framework; (b) proposed Zn2+ diffusion pathway in ZnMn2O4 spinel without and with Mn vacancies 59; (c) GITT profiles and calculated Zn2+ diffusion coefficient of ZMO/C and ZMO + C electrodes; (d) crystal structure, (e) charge/discharge profiles and(f) corresponding cycle performance of MgMn2O4 at 500 mA·g-1 136.
图16 (a) CaSO4·2H2O SEI膜覆盖Ca2MnO4的结构形成机理;Ca2MnO4与α-MnO2的(b) Mn溶解率以及(c)活化能对比138Fig.16 (a) The formation mechanism of unique structure of CaSO4·2H2O SEI layer-coated Ca2MnO4;comparison of (b) manganese dissolution rate and (c) activation energy of Ca2MnO4 and α-MnO2 138.
3.2.4 密排六方结构
Akhtenskite型 MnO2(ε-MnO2)是 由 MnO6和YO6八面体(Y为空位)共面组成的亚稳相,其中Mn4+随机占据密排六方晶格中50%的八面体位点37。Wang等139将其作为锌离子电池正极并实现了超500次的稳定循环,在0.1 A·g-1的电流下可获得221 mAh·g-1的比容量。但当电流密度增加至4.3C时,容量却不足100 mAh·g-1。因此,ε-MnO2的储锌性能还有待优化。近日,Lu等137报道了一种La3+和Ca2+共嵌的ε-MnO2海胆状纳米结构(LCMO),这种结构为Zn2+的嵌入提供了丰富的活性位点。La3+和Ca2+的共嵌使ε-MnO2晶格间隙扩大,锌离子扩散系数相比未掺杂时提高了一个数量级。掺杂后电荷转移阻抗(Rct)从198.1 Ω降到了29.8 Ω,说明在La3+和Ca2+的调控下ε-MnO2的带隙得以缩小,导电性能得到提升。对比单离子掺杂实验结果得知,Ca2+对提升结构稳定性的作用较大,而Ca2+和La3+皆能一定程度提高ε-MnO2的放电比容量。基于这些优点,在0.2和1.6 A·g-1的电流密度下,LCMO能分别输出297.3和161 mAh·g-1的比容量,并且循环200次后仍有76%的容量保持率,较未掺杂时提升了29%。
3.2.5 其他结构
针对锰基材料的溶解和动力学缓慢等问题,本课题组提出了一种钙离子预嵌的锰基氧化物(Ca2MnO4)138。在首次充电过程中,预嵌的Ca2+从Ca2MnO4中脱出并在表面原位生成单一组分的CaSO4·2H2O界面保护膜(图16a),在Ca2+预嵌提升本征结构稳定性的基础上,界面保护膜的生成进一步抑制了锰的溶解(图16b)。理论计算和实验结果表明,CaSO4·2H2O界面膜还具有降低阻抗、改善界面、减少活化能(图16c)的作用,有效地促进了Zn2+的嵌入与脱出,进一步提高了电池的循环性能和倍率性能,使其能在1 A·g-1的电流密度下稳定循环1000次。这种原位电化学生成界面保护膜的方法为今后发展高稳定性的水系电池开拓了新视野。
3.3 阴离子预嵌
3.4 有机分子预嵌
随着充放电过程中锰的不断溶解以及Zn2+、H+和水分子的嵌入,各种结构的MnO2最终都将转变为层状结构24,115,因此从理论上来说,为了避免相变对电极结构的破坏,直接选用层间距较大的δ-MnO2作为Zn2+/H+的宿主材料更为合适。但当水合阳离子嵌入过多时,层状结构同样容易发生崩塌,导致容量迅速下降116。为了提高δ-MnO2的容量和循环稳定性,Xia等134通过简单的有机/无机界面反应合成了一种聚苯胺(PANI)预嵌的层状MnO2(图17d)。PANI的预嵌显著提高了MnO2的电导率和扩散动力学,并有效地抑制了锰的溶解。该材料在200 mA·g-1的电流密度下可以贡献280 mAh·g-1的比容量并稳定循环200次,容量利用率达到了Mn4+/Mn3+单电子反应理论容量(308 mAh·g-1)的90%。
采用导电聚合物包覆、复合的手段虽然可以有效提高锰基材料的导电性,但往往会带来一些附加问题,如离子扩散受阻、活性物质与导电网络联结性较差等。为此,Xue等135以十二烷基硫酸钠(SDS)为抑制剂,合成了一种聚吡咯(PPy)水平外延生长的二维MnOx纳米层。这种结构具有以下几点优势:柔性的PPy分子链缓解了Zn2+嵌入/脱出过程中的体积膨胀,有效地提升了结构稳定性;Mn、N原子间的相互作用降低了MnOx与PPy间的界面阻抗(图17e),使整个电极具有良好的导电性;MnOx的高比表面积使其与电解液接触充分,活性物质的利用率较高。因此,PPy/MnOx纳米层在1C的倍率下可以贡献408 mAh·g-1的高比容量,以5C的倍率循环3000次后容量保持率仍高达78%。

图17 MnO2@VMG和P-MnO2-x@VMG中(a) P 2p和(b) O 1s的XPS谱图以及(c)两种电极的倍率性能对比133;(d)聚苯胺(PANI)预嵌入MnO2纳米层的形成原理图134;(e) MnOx/PPy,MnOx,PPy的交流阻抗谱135Fig.17 XPS spectra with (a) P 2p, (b) O 1s, and (c) rate performances of MnO2@VMG and P-MnO2-x@VMG 133;(d) schematic illustration of expanded intercalated structure of polyaniline (PANI)-intercalated MnO2 nanolayers 134;(e) EIS patterns of MnOx/PPy, MnOx and PPy 135.
4 总结与展望
水系锌离子电池具有高安全、低成本、高能量、环境友好等优点,在大规模储能领域表现出了良好的应用前景。在众多正极材料中,钒、锰基化合物的储锌性能较为优异,近年来引起了广泛的关注。虽然这两种材料具有较高的比容量和合适的嵌锌电位,但电导率低、结构稳定性差、反应动力学行为缓慢等问题仍然限制了其走向实际应用。在本综述中,我们重点关注客体预嵌策略对钒、锰基正极材料的电化学性能的影响,并对其研究进展进行了总结。总体而言,阴阳离子、水和有机分子等客体可以作为“支柱”有效地稳固钒、锰基宿主的晶体结构,抑制其组分的溶解,增强电极的导电性和反应动力学行为,从而提高电池的循环性能和倍率性能。然而现阶段单靠客体预嵌策略对正极材料性能的提升是十分有限的,通常还需要结合缺陷工程、纳米结构调控、材料复合等优化手段,并且许多问题并没有从根本上得到解决。另外,客体是否主要通过扩大正极宿主的层间距来提升电化学性能这一点仍存在着争议,其真正的强化机制还需要更深入的研究和讨论。
为了在现有的基础上对水系锌离子电池的电化学性能进行突破,未来仍需从以下几个角度对钒、锰基正极材料进行深入挖掘:
(1)电化学机理研究。目前报道的钒、锰基正极材料储能机理涉及嵌入机制、转化机制、溶解-沉积机制等,弄清其储能机理,才可以针对性改善其电化学性能。对许多钒、锰基化合物而言,质子(H+)与Zn2+的共嵌是获得优异电化学性能的重要原因。从动力学角度考虑,H+的嵌入比Zn2+更加容易,但H+浓度的变化会导致副反应产物如Zn4(OH)6SO4·nH2O在正极表面的生成与消失。这种机制易造成电解液的消耗以及界面阻抗的增加,并且在实际应用中,Zn4(OH)6SO4·nH2O的脱落还会增加电化学行为的不可逆性,使得容量衰减较快,循环寿命减短。然而目前关于副产物的研究较少,其对整个储能体系的影响尚不明确。因此,未来需结合原位表征技术和理论计算方法,探究正极的微观结构、组分、界面与其电化学行为之间的联系,寻找到限制电池性能的内在原因,并对此进行针对性优化。
(2)抑制钒、锰基材料在水系电解液中的溶解。正极材料在水溶液中的溶解问题,是除Zn2+扩散动力学缓慢之外限制锌离子电池性能的重要因素。一方面,正极的溶解会造成活性物质的损失,使得容量大大降低;另一方面,已溶解的金属离子易在负极界面生成钝化膜,降低锌的活性。钒基材料的溶解一般与充放电时间长短有关,具体体现在小电流循环时容量的迅速衰减以及大电流循环时容量的相对稳定。锰的溶解主要是由循环过程中正极的结构变化、Mn3+的歧化反应以及可溶性Mn2+的生成引起的。如前文所述,预嵌客体可以通过形成层间支柱或构建隧道晶体结构等方式,形成更稳定的V-O、Mn-O键,减少正极在Zn2+或H+嵌入/脱出时发生的结构劣变,降低活性物质的溶解,从而提高循环稳定性。但从表1和表2可以得知,许多客体预嵌钒、锰基材料的长循环寿命都是在2 A·g-1及以上的大电流条件下实现的,小电流下材料溶解造成的容量衰减问题依然严重。目前还可以通过在电解质中添加相应的盐、对电极进行表面包覆以及采用固态电解质的方法对溶解问题进行抑制。然而这些方法的成本较高,不利于实现商业化生产。因此,如何从正极的本征结构出发,在保证低成本的同时降低其在水溶液中的溶解率,提高循环稳定性,未来仍需要更深入的研究。
(3)开发高电压、高容量的新型钒、锰基材料。水系锌离子电池受到析氧反应的限制,其工作电压通常低于2 V,这也是阻碍其走向实际应用的最主要原因。虽然已有研究通过引入PO3-、F-等阴离子或采用“盐包水”型高浓度电解液来拓宽电压窗口,但其容量较低,能量密度仍然不够理想。今后仍需对钒、锰基材料进行电子结构的调控,提高其嵌锌电位,并结合负极、电解液的优化以及电池结构的设计,突破一些技术屏障,在已有高比容量的基础上实现高工作电压,以此获得更高能量密度的水系锌离子电池。
(4)开发高负载量、高性能的钒、锰基正极。钒、锰基材料自身的电导率较低,粘附性较差,在正极的涂覆过程中通常需要添加导电剂和粘结剂来保持一定的电化学性能。因此,正极活性物质的负载量以及电池整体的能量密度都受到了极大的限制。在近几年的报道中,大多数性能优异的正极材料其负载量都落于1–3 mg·cm2之间。然而在实际应用中,为了保证能量密度,往往需要对正极进行更高量级的负载,这就对正极在高负载情况下的活性物质利用率提出了要求。为了实现水系锌离子电池的商业化应用,未来还需开发自支撑、高电导率的钒、锰基材料,降低添加剂的使用量,提高正极活性物质的负载量和利用率,并优化其电化学性能。
猜你喜欢
山东冶金(2019年5期)2019-11-16 09:09:12
重型机械(2019年3期)2019-08-27 00:58:44
焊接(2016年9期)2016-02-27 13:05:22
电源技术(2016年2期)2016-02-27 09:04:59
中国资源综合利用(2016年7期)2016-02-03 03:00:19
新疆钢铁(2015年2期)2015-11-07 03:27:52
电源技术(2015年12期)2015-08-21 08:58:20
风能(2015年8期)2015-02-27 10:15:12
风能(2015年5期)2015-02-27 10:14:46
应用化工(2014年1期)2014-08-16 13:34:08
