
应用ABEEM方法计算含金属离子Ga3+蛋白的电荷分布
2021-04-02 13:09:52卢丽男杨忠志
辽宁师范大学学报(自然科学版) 2021年1期
卢丽男, 杨忠志
(辽宁师范大学 化学化工学院,辽宁 大连 116029)
金属镓(Ga)作为第三主族元素,其表观存在形式常为Ga3+,为高价态的金属离子.在1931年研究表明镓离子具有抗菌活性.镓制剂抗菌活性的研究不断地拓展和深入,表明了不同种类的镓化合物都具有抗菌活性,并且尝试表征和开发含镓的抗菌药物[1-2].金属离子和蛋白的相互作用的研究和模拟得到了迅速的发展.现阶段对于二价金属离子和蛋白的相互作用研究较多,例如Zn2+、Cu2+和Mg2+离子体系[3-8].对于高价态的金属离子Ga3+与蛋白的研究较少,高价态的金属离子对于理论模拟和计算具有很大的挑战[9].对于含有金属离子蛋白大分子体系的研究,量子化学方法目前计算的原子数目非常有限.金属蛋白的相关分子力场研究得到了迅速的发展,其中,大分子体系的电荷分布十分重要.杨等人根据密度泛函理论(DFT)和电负性均衡原理(EEM)发展了ABEEMσπ计算电荷方法(简称为ABEEM方法),可用于计算大分子体系的电荷分布[10-16]. ABEEM方法将分子电荷分解到了原子区域、σ键区域、π键区域和孤对电子区域.每个区域的选取都有实际的物理意义,而且点电荷会随着周围环境而浮动变化,能够很好地体现体系的静电极化现象.ABEEM方法引入了一个能够反映氢键的特殊效应的氢键拟合函数,从能量和成键角度更合理地描述氢键相互作用,能够快速、合理地得到大分子体系的电荷分布.
首次开发ABEEM方法应用于含金属离子Ga3+蛋白体系的研究,金属离子和蛋白之间的相互作用处理为成键模型,能够很好地稳定金属离子和周围配体的配位.并且体系采用电荷整体守恒,能够充分反映出原子之间的相互作用和电荷转移,能够很好地反映体系的极化现象.为高价态的金属离子和蛋白的相互作用做出开创性的研究,为研究大分子体系电荷分布提供了一个可靠的新方法,为建立分子力场进行力学模拟打下基础.
1 计算方法
1.1 ABEEM方法
体系基态能量用ABEEM方法表示为
(1)
根据密度泛函理论电负性的定义,由方程(1)就可以定义分子中的任意一区域(原子、化学键、孤对电子,以及双键中的σ和π区域)的有效电负性,表达为方程(2):
(2)
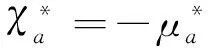
1.2 模型分子
从蛋白质晶体数据库里截取出含有金属离子Ga3+蛋白模型,通过Gaussian View 5.0软件给模型分子初步加上氢,使用Gaussian 09程序包的M06-2X/6-311+G(d)的方法,保持晶体结构的重原子坐标不变,对氢进行优化,最终确定整个模型分子的结构.用HF/STO-3G计算得到的模型分子的电荷分布,作为ABEEM方法计算电荷的参考标准.选择最小的STO-3G基组计算的电荷作为参考标准,并不是因为它的计算消耗小,而是因为它的物理意义.较低等的基组更能给出接近实际应用的电荷分布,原因是高等的方法和基组,使位于原子上的扩散基函数可能在某种程度上覆盖了其他相邻原子的区域,结果在Mulliken布居分析中会比小的基组方法高估该原子的布居数.具体的比较数据和说明参见文献[13].
截取得到11个含金属离子Ga3+蛋白模型分子,共分为4类.还截取得到4个含金属离子Ga3+蛋白的大分子体系,分别为1CFWJ-large(含有155个原子)、2KT4J-large(含有229个原子)、3QUG1-large(含有238个原子)、6J2S1-large(含有210个原子),进一步验证了ABEEM方法的电荷参数的正确性、合理性和可转移性.具体模型分子如图1所示.

图1 模型分子(a)4类金属离子Ga3+蛋白模型(b)4个金属离子Ga3+蛋白大分子模型Fig.1 Model molecules (a) 4 types of Ga3+ protein model (b) 4 Ga3+ protein large molecular model
2 结果与讨论


表1 ABEEM方法计算含金属离子Ga3+蛋白模型电荷相关参数
利用以上调节得到的ABEEM方法的相关参数计算了4类共11个金属离子Ga3+蛋白模型分子的电荷分布,同时与HF/STO-3G方法的计算结果进行对比.从表2中可以看到两种方法下计算的电荷结果的线性相关方程的斜率A接近于1.00,截距接近于0,线性相关系数R大部分在0.98以上(3QUG1为0.97),标准偏差S接近于0.ABEEM方法计算电荷的结果与HF/STO-3G方法有很好的相关性,说明ABEEM方法计算电荷的合理性和正确性.

表2 ABEEM方法计算金属离子Ga3+蛋白模型的电荷分布与HF/STO-3G结果的相关性
在得到的ABEEM方法的电荷参数没有进行任何改动的前提下计算4个金属离子Ga3+蛋白大分子模型分子的电荷分布.ABEEM方法几秒钟就可以计算完成含有几百个原子的模型分子的电荷分布.与HF/STO-3G计算得到的结果进行了对比分析,具体参见图2.从图2中可以看到两种方法下的计算电荷结果的线性相关方程的斜率A接近于1.00,截距接近于0,线性相关系数R在0.95以上,标准偏差S接近于0.ABEEM方法计算4个金属离子Ga3+蛋白大分子模型分子的电荷分布的结果可以和HF/STO-3G方法计算结果有很好的相关性,验证了ABEEM方法可以准确地计算大分子体系的电荷分布,电荷参数具有很好的可转移性.

图2 4个Ga3+蛋白大分子模型分子的ABEEM和HF/STO-3G方法的电荷分布相关图(A)1CFWJ-large;(B)2KT4J-large;(C)3QUG1-large;(D)6J2S1-largeFig.2 Comparison of charge distributions of 4 Ga3+ protein macromolecular model molecules obtained from the ABEEM and the HF/STO-3G methods(A)1CFWJ-large;(B)2KT4J-large;(C)3QUG1-large;(D)6J2S1-large
高价态的Ga的电荷分布对整个含金属离子Ga3+蛋白来说尤为重要,其电荷分布的合理性和正确性直接影响着整个研究体系性质的模拟质量.ABEEM和HF/STO-3G两种方法计算得到的上面所有模型分子(共15个)的Ga的电荷分布展现在图3中.金属离子Ga3+在和蛋白相互作用后,从周围的配体中得到电子,由原来表观带3个正电荷变成更小的正电荷.Ga原子的电荷分布范围很大,HF/STO-3G计算得到的镓原子的电荷分布在0.278 4e到1.615 8e,ABEEM方法计算得到的镓原子的电荷分布在0.279 2e到1.573 9e.浮动范围略小于HF/STO-3G的结果.镓原子的电荷这么大的浮动范围,而ABEEM模型可以很好反映镓的电荷变化.从每个模型分子的镓的电荷分布情况可以看出,ABEEM模型计算的大部分模型分子的镓电荷分布略高于HF/STO-3G的计算结果,两种方法计算的镓的电荷平均绝对偏差为0.102 5e.ABEEM方法能够很好地反映体系的电荷转移,例如在1CFW1结构中,半胱氨酸的残基本身电荷为0,而和金属离子Ga3+相互作用后,正三价的Ga3+有强烈的吸引电子的能力,使4个配位的半胱氨酸的残基分别带有正电荷.ABEEM方法计算得到的4个配位的半胱氨酸残基的电荷为0.718 9e、0.630 6e、0.683 7e和0.684 2e,HF/STO-3G方法计算的结果为0.677 1e、0.680 0e、0.661 7e和0.697 0e.两种方法的计算结果十分符合.

图3 金属离子Ga3+蛋白模型分子中Ga的ABEEM方法和HF/STO-3G的电荷分布图Fig.3 Comparison of charge distributions of Ga in protein macromolecular model molecules obtained from the ABEEM and the HF/STO-3G methods
3 结论与展望
扩展ABEEM电荷方法应用范围,首次设计含金属离子Ga3+蛋白模型,Ga3+与周围配体为成键模型,并和配体之间有电荷转移,电荷守恒条件为整个分子守恒.ABEEM电荷模型的电荷是浮动的,很好地体现了金属离子和配体的相互作用和极化.通过大量的计算分析工作协调优化确定ABEEM电荷方法的电荷参数,计算了11个金属离子Ga3+蛋白模型分子和4个金属离子Ga3+蛋白大分子模型的电荷分布,并和HF/STO-3G的计算结果进行了对比.两种方法的电荷结果有很好的相关性,线性相关方程的斜率A接近于1.00,截距接近于0,线性相关系数R在0.95以上,标准偏差S接近于0.重点比较了15个模型的金属离子Ga3+的电荷,以及和周围配位基团的电荷转移情况.以上研究验证了ABEEM方法可以合理地计算含金属离子Ga3+蛋白模型分子的电荷分布,电荷参数具有很好的可转移性,将为高价态金属离子和蛋白的相关研究给出重要的参考,为分子动力学模拟打下基础.
猜你喜欢
物理通报(2024年4期)2024-04-09 12:41:28
中学生数理化·中考版(2021年10期)2021-11-22 07:26:40
中学生数理化(高中版.高考理化)(2020年10期)2020-10-27 03:07:02
无线互联科技(2018年6期)2018-06-25 07:34:40
浙江大学学报(理学版)(2016年6期)2016-12-15 03:15:03
分析测试学报(2015年3期)2016-01-13 06:18:12
物理化学学报(2015年7期)2015-12-30 12:13:08
新高考·高一物理(2015年6期)2015-09-28 20:10:57
原子与分子物理学报(2015年3期)2015-07-13 03:39:22
湖南师范大学学报·自然科学版(2014年4期)2014-10-23 12:05:20
