
GCH1基因剪切突变致多巴反应性肌张力障碍1例
2021-03-31 09:33:36任淑红张晓燕
中国妇幼健康研究 2021年3期
董 畅,任淑红,张晓燕
(保定市儿童医院神经内科,河北 保定 071000)
多巴反应性肌张力障碍(dopa-responsive dystonia,DRD)是一组基因相关的遗传性疾病,与左旋多巴胺合成代谢通路上的酶活性缺陷有关,进而出现进行性肌张力障碍。本病患病率较低,以女性多见,女性为男性的2~4倍。DRD主要是由于三磷酸鸟苷环化水解酶1(guanosine triphosphate cyclohydrolase 1,GCH1)基因和酪氨酸羟化酶(tyrosine hydroxylase,TH)基因突变所致,不同基因缺陷所致临床表现具有明显异质性,故该病的诊断存在较多困难。本文报道1例GCH1基因内含子突变所致DRD,从而进一步探讨GCH1基因对该病的影响。
1临床资料
患儿,女,5岁,主因走路姿势异常、易摔跤3年余,于2017年2月就诊于我院。患儿自1岁3个月开始学步走路后,无明显诱因出现走路姿势异常,动作笨拙,步态不稳,易摔跤,行走速度慢,可自行上下楼梯,不会双足及单足跳,家长未予特殊治疗,上述症状进行性加重,似有晨轻暮重的特点,患儿语言及智力发育正常,无肢体疼痛、无力,无感觉异常及尿便障碍。自发病以来,患儿精神状态良好,体力情况良好,食欲食量良好,睡眠情况良好。既往史无特殊,否认传染病接触史,本患儿为第2胎,足月剖宫产,围产期(-),生长发育史同正常幼儿,否认家族遗传史。查体:体重27.00kg,神志清,精神反应可,计算力、理解力、记忆力正常,定向力正常,可正常对答,对答切题,吐字清晰,角膜K-F环(-),颅神经查体阴性,四肢肌力正常,肌张力正常,双侧膝腱反射活跃,踝阵挛(+),巴氏征(+),Gower征(-),其余神经系统查体(-)。门诊辅助检查:血常规、肝肾功能、心肌酶、电解质、血沉、抗核抗体谱、叶酸、维生素B12、血尿筛查、肌电图均正常。骨盆正位片、颈腰椎核磁、头颅CT及核磁均未见明显异常。异常辅检:乳酸3.10mmol/L,血氨111.56μmol/L,呈升高,复查后恢复正常。双侧髋膝关节彩超示双侧股骨颈前间隙增宽并伴积液。骨密度检测示中低水平。2017年2月该患儿及家属完善家系基因检测(见表1及图1),受检者及其父亲均发现GCH1基因c.542-2A>T(编码区第542号核苷酸前内含子中倒数第2位核苷酸由A变为T)的杂合核苷酸变异,其母亲、大姐和弟弟未发现变异。
患儿及其父亲发现的GCH1基因c.542-2A>T属于剪切变异,与该变异同一位点的c.542-2A>C的致病性已经有文献报道[1],且与DRD相关,该变异不属于多态性变化,在人群中发生的频率极低。该基因突变经MutationTaster软件预测蛋白功能损伤为可能致病,变异分级评分为可能致病,且该变异与疾病的相关性在HGMD数据库中有收录[1]。患儿予以美多芭31.25mg/次,2次/日,口服治疗后,用药1周患儿动作笨拙较前缓解,2个月后走路姿势逐渐正常,4个月后可双足跳,无智力及生长发育落后表现。患儿目前体重32.00kg,美多芭用量为62.50mg,2次/日,可以双足跳,不会单足跳,至今未出现症状反复或不良反应。
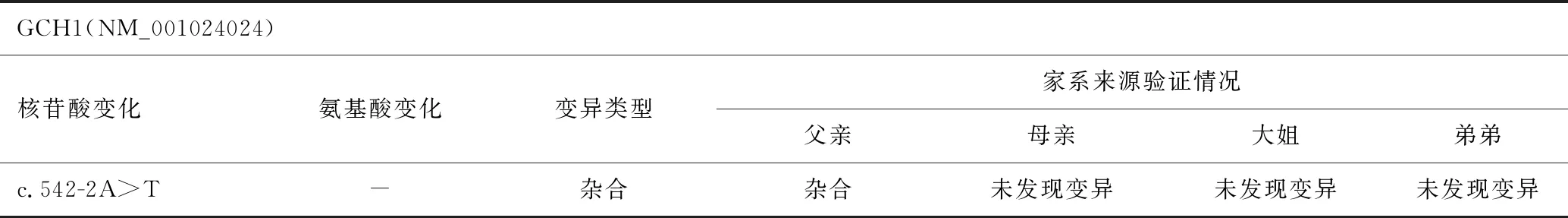
表1 患儿家系GCH1基因变异及类型
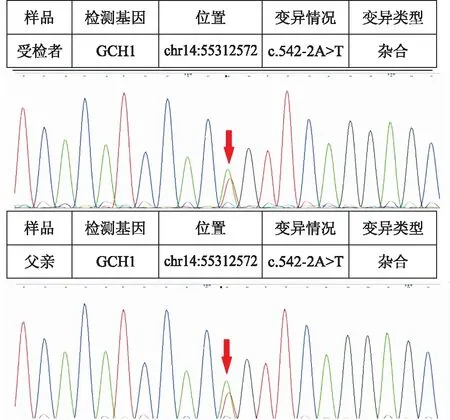
图1 患儿及父亲GCH1基因突变位点
2讨论
2.1多巴反应性肌张力障碍的临床特点
DRD属于一种遗传性锥体外系疾病,是由于多巴胺合成不足导致纹状体功能障碍[2],绝大多数在儿童及青年期起病,具有不完全外显率和高变异性的特点。通常的表型为早发孤立性肌张力障碍,主要发生在下肢,症状具有昼夜波动性,对低剂量左旋多巴反应剧烈而持续[3],亦称Segawa病。典型的临床表现为双下肢姿势异常,会出现尖足行走或马蹄内翻足。疾病症状的波动性除晨轻暮重表现,有些在疲劳或感染情况下加重,在休息或睡眠后减轻或消失。非典型临床特征包括步态蹒跚、全身性肌张力减退和近端肢体无力。越来越多的学者认为焦虑、抑郁及强迫症等非运动症状也应归为GCH1缺乏引起的临床表现,因其缺乏导致四氢生物碟呤(BH4)合成不足及5-羟色胺低水平,引起相关精神症状[4]。治疗方面,外国一项研究将DRD患者步态、姿势等参数量化,根据参数定量分析左旋多巴治疗后效果,结果显示下肢运动参数几乎全部得到明显改善,提示左旋多巴剂量个体化治疗效果显著[5]。据统计,DRD在国内外发病率不高,约为5/1000万,因常有报道如腱反射亢进、踝关节阵挛等患者被误认为脑瘫,故其患病率高于发病率,需注意鉴别。本病例为学龄前期患儿,以双下肢走路姿势异常起病,表现为步态不稳、动作笨拙,症状有晨轻暮重特点,患儿病史偏长,但予以美多芭治疗后临床症状很快恢复且无运动并发症,符合经典型DRD的表现。
2.2 GCH1基因突变形式
GCH1基因位于人类第14号染色体上(14q22.1-22.2),包含6个外显子及2个内含子。DRD遗传方式分为常染色体隐性遗传(AR-DRD)和常染色体显性遗传(AD-DRD)两种。常染色体显性遗传DRD主要由三磷酸鸟苷环化水解酶1基因缺陷引起,为临床典型病例的50%~60%,而隐性遗传主要由酪氨酸羟化酶(TH)基因缺陷引起[6]。GCH1基因编码的三磷酸鸟苷环化水解酶1是参与BH4合成的关键酶,而BH4是参与多巴胺合成过程中的限速酶,BH4的减少最终会导致多巴胺等神经递质的减少甚至缺乏,进而出现一系列临床症状。目前根据国内外文献描述,共发现GCH1基因编码区的228种突变[7],且其突变率会逐渐升高。DRD的GCH1基因异常包括错义突变、无义突变、移码突变和剪切突变。约15%的GCH1基因检测阴性的患者可能存在深部内含子突变异常,或尚未明确的基因突变形式[8]。既往研究报道显示,大部分GCH1基因突变集中于编码蛋白质的外显子区域,本研究中患儿GCH1基因c.542-2A>T属于内含子部位的剪切变异,该变异评级为可能致病,目前该位点国外文献已有收录,但国内尚未出现相同位点突变报道。
2.3临床遗传异质性
有研究表明男性可能在同家系中仅表现为异常基因的携带者,而女性则可出现DRD临床症状[9]。国外研究发现通常无症状携带者GCH1的活性下降至30%~40%,当GCH1的活性下降到正常水平的20%以下会出现相应临床症状。本例患儿及其父亲均为GCH1基因内含子相同部位的剪切突变,而家族中其他人员未检测到基因突变,父亲无该病临床表现,符合该病临床表现外显率可能受到性别或年龄的影响[10]。患儿经美多芭治疗后临床症状有所好转,可间接证明其致病性,据ACMG指南关于变异分级评分为可能致病性变异,结合患儿的临床表现,支持致病证据,推测内含子剪切位点突变导致成熟RNA发生变异致翻译的多肽链结构异常而致病。总之,DRD患儿的临床表现复杂多样,在诊断不明的前提下尽早完善基因检测,可以有效避免漏诊误诊,或可检测GCH1的活性判断干预时间及治疗效果,有效改善预后。
猜你喜欢
英语世界(2023年6期)2023-06-30 06:29:10
内蒙古师范大学学报(自然科学汉文版)(2021年3期)2021-06-01 08:10:30
中国生殖健康(2020年2期)2021-01-18 02:51:26
趣味(数学)(2020年4期)2020-07-27 01:44:16
支部建设(2020年15期)2020-07-08 12:34:32
生物工程学报(2019年6期)2019-07-10 08:38:38
生物学通报(2019年1期)2019-02-15 16:33:43
生物学通报(2018年12期)2018-10-10 06:52:36
小学生导刊(2018年13期)2018-06-29 03:49:00
百科知识(2015年18期)2015-09-10 07:22:44
