
谷氨酸-谷氨酰胺循环异常与孤独症谱系障碍研究进展
2021-03-30 07:24袁启锋姚宝珍
生物技术进展 2021年2期
袁启锋, 姚宝珍
武汉大学人民医院儿科, 武汉 430060
孤独症谱系障碍(autism spectrum disorder,ASD)是一种多病因神经发育性疾病,人群患病率约为1%,其中男性患病率约为1∶42,是女性患病率的4倍[1]。其临床核心症状为社会交往障碍、重复刻板样动作和狭窄兴趣,除核心症状外,ASD患者还常表现有焦虑、抑郁、多动、冲动、易激惹、智力障碍、注意力缺陷、睡眠障碍、癫痫、便秘等伴随症状[2]。自1940s首次描述ASD[3],科学界对其病因进行了广泛探讨,包括炎症、自身免疫、基因异常等,但具体发病机制仍未明了。
在哺乳动物中枢神经系统(central nervous system,CNS)中,谷氨酸是最主要的兴奋性神经递质,也是一种潜在的神经毒素,其引起的兴奋毒性可能导致神经细胞的死亡。为了保持突触传递的敏感性,谷氨酸必须被及时清除,其中,星形胶质细胞的摄取是保持胞外谷氨酸浓度稳定的最主要途径[4]。星形胶质细胞和神经元之间谷氨酸-谷氨酰胺的代谢偶联(图1),可以防止过量的谷氨酸扩散到周边的神经元上,进而避免了神经元的过度兴奋,对神经元起到保护作用。谷氨酸能神经元是哺乳动物大脑皮层最主要的神经元。在谷氨酸能神经元中,谷氨酰胺(glutamine,Gln)经谷氨酰胺酶(glutaminase,GLS)催化为谷氨酸(glutamate,Glu),Glu与N-甲基-D-天冬氨酸受体(N-methyl-D-aspartic acid receptor,NMDAR)、α-氨基-3-羟基-5-甲基-4-异恶唑丙酸受体(α-amino-3-hydroxy5-methyl-4-isoxazole-propionic acid receptor,AMPAR)、红藻氨酸受体(kainate receptor,KAR)、代谢性谷氨酸受体(metabotropic glutamate receptors,mGluRs)结合,形成兴奋性电位。星形胶质细胞可通过膜上谷氨酸转运体(glutamate transporter, GLT)摄取突触间隙Glu,在谷氨酰胺合成酶(glutamine synthetase,GS)作用下转化为Gln,后者被星形胶质细胞释放,可再次进入Glu能神经元或者γ-氨基丁酸(γ-aminobutyric acid,GABA)能神经元。在GABA能神经元,Glu被谷氨酸脱羧酶(glutamic acid decarboxylase,GAD)催化为GABA,与Glu能神经元相类似,GABA与GABA受体结合,产生抑制性电位,被星形胶质细胞经GABA转氨酶(GABA transporter,GAT)摄取的GABA通过三羧酸循环(tricarboxylic acid cycle,TAC),重新转化为Glu和Gln,从而被再次利用[5]。一系列研究表明,Glu与GABA在ASD患者不同脑区表达水平不同[6-8],提示谷氨酸-谷氨酰胺循环异常可能为ASD发生的核心机制。因此,本文围绕谷氨酸-谷氨酰胺循环与ASD的关系进行综述,重点探讨炎症、自身免疫、基因异常等ASD经典致病因素与该循环异常之间的联系,以期为孤独症谱系障碍分型和治疗提供一种新思路。

图1 谷氨酸-谷氨酰胺循环模式Fig.1 Glutamate-glutamine cycle mode
1 谷氨酸-谷氨酰胺循环异常与ASD
1.1 Glu、GABA水平与ASD
当前,关于ASD患者Glu、GABA水平的研究结论并不一致(表1),这可能是由于研究对象在年龄、性别、受试者数量等因素方面存在较大的差异且所取的生物学样本不同[13]。后续研究可以通过标准化这些因素来改进,并且利用Glu/GABA比值来代替单个氨基酸水平,以更好地反映神经兴奋/抑制平衡。由于Glu、GABA在ASD发病机制和治疗中的潜在作用,监测其在体液乃至唾液、尿液和粪便中的浓度变化对ASD患者的早期诊断和干预也很重要;此外,有研究提示GABA可能是一种灵敏度和特异度均较高的ASD血清标志物[14]。
1.2 NMDAR与ASD
NMDAR是一种配体门控兴奋性离子通道受体,在神经元兴奋性毒性和突触可塑性方面发挥重要作用。NMDAR广泛分布于大脑,由GluNR1~GluNR3 3个亚基构成:GluNR1含甘氨酸结合位点;GluNR2有A、B、C、D 4种亚型,含Glu结合位点;部分NMDAR还含有GluNR3亚基,可抑制该受体功能。静息状态时,NMDAR钙离子(Ca2+)通道由水合镁离子(Mg2+)堵塞。当突触前膜释放Glu等神经递质,激活AMPAR等受体,突触后膜去极化,Mg2+被移除,此时甘氨酸结合GluNR1亚基与Glu结合GluNR2亚基共激动NMDAR,Ca2+通道开放,Ca2+进入神经元细胞质。当突触间隙Glu浓度异常升高,NMDAR过度激活时,神经元胞质内钙超载,活化相关代谢酶、生成氧自由基、破坏细胞能量产生,进而损伤神经元[15]。NMDAR过度激活所导致的神经元兴奋性毒性损伤可能与ASD发病相关。研究发现,ASD患者血清甘氨酸浓度更高[16],同样,丙戊酸(valproic acid,VPA)诱导的ASD模型小鼠前额叶皮层GluNR2B亚基水平也较正常组高[17],提示ASD患者NMDAR功能增强。而使用NMDAR拮抗剂MK-801(出生后6~10 d给药),可改善VPA模型ASD鼠社会交往障碍和重复刻板样动作[18],但MK-801(0.03 mg·kg-1)也会损害正常小鼠社会交往功能[19],这可能与MK-801剂量有关,剂量不同,它的生物效应可能截然相反[20]。D-环丝氨酸,作为甘氨酸结合位点相关的NMDAR部分激动剂,可作用于NMDAR,进而促进AMPAR移除来改善ASD模型鼠(出生后28 d给药)社会交往障碍,且不同于Glu直接激活NMDAR,D-环丝氨酸并不会导致兴奋性神经毒性[21]。NMDAR的激活需AMPAR参与,故D-环丝氨酸最终可能通过降低VPA暴露后代的NMDAR功能来改善ASD症状,当然这需要进一步试验来验证。此外,ASD鼠NMDAR功能可能与小鼠年龄密切相关。Shank2-/-小鼠是经典的ASD模型,研究发现,这种小鼠在出生早期(出生后15 d内)表现为NMDAR功能亢进,后期则表现为NMDAR功能减退,早期使用NMDAR拮抗剂——美金刚,可有效缓解小鼠社会交往障碍,而早期使用D-环丝氨酸或后期使用美金刚则均无明显效果[22]。那么,VPA诱导的ASD模型鼠甚至临床ASD患者NMDAR功能是否随年龄有这种类似转变,仍需进一步深入探究。

表1 谷氨酸-谷氨酰胺循环异常与ASD发病相关研究Table 1 Study on the relationship between abnormal glutamate-glutamine cycle and ASD
2 炎症与ASD
炎症是机体的一种固有免疫方式,正常机体促炎细胞因子/抗炎细胞因子处于平衡状态。肥胖与促炎细胞因子/抗炎细胞因子失衡相关。值得注意的是,与正常人群相比,ASD患者常有肥胖表现[23]。提示ASD可能与炎症相关[24-26]。进一步研究发现,肿瘤坏死因子-ɑ(tumor necrosis factor-ɑ,TNF-ɑ)mRNA表达水平与ASD患者社会行为异常有关,与患者其他核心症状相关性则未得到证实[27]。
炎症可通过影响谷氨酸-谷氨酰胺循环导致ASD发生(图2)。在ASD动物模型中,促炎细胞因子/抗炎细胞因子失衡与谷氨酸-谷氨酰胺循环异常并存[25]。星形胶质细胞在谷氨酸-谷氨酰胺循环中具有重要作用,神经炎症可致GLT-1表达上调和GS表达下调,进而影响星形胶质细胞的数量和功能,导致其对Glu摄取能力下降,过度激活NMDAR,造成神经元兴奋性毒性损伤[28-29]。为探究ASD患者神经炎症与谷氨酸-谷氨酰胺循环异常之间的因果关系,El-Ansary等[30]对20名ASD患儿血清样本进行多元回归分析,认为ASD患儿血清Glu升高87%归因于神经炎症。进一步研究还发现,神经炎症可激活犬尿氨酸途径(kynurenine pathway,KP),该途径代谢产物喹啉酸过度激活NMDAR导致ASD发生[31]。
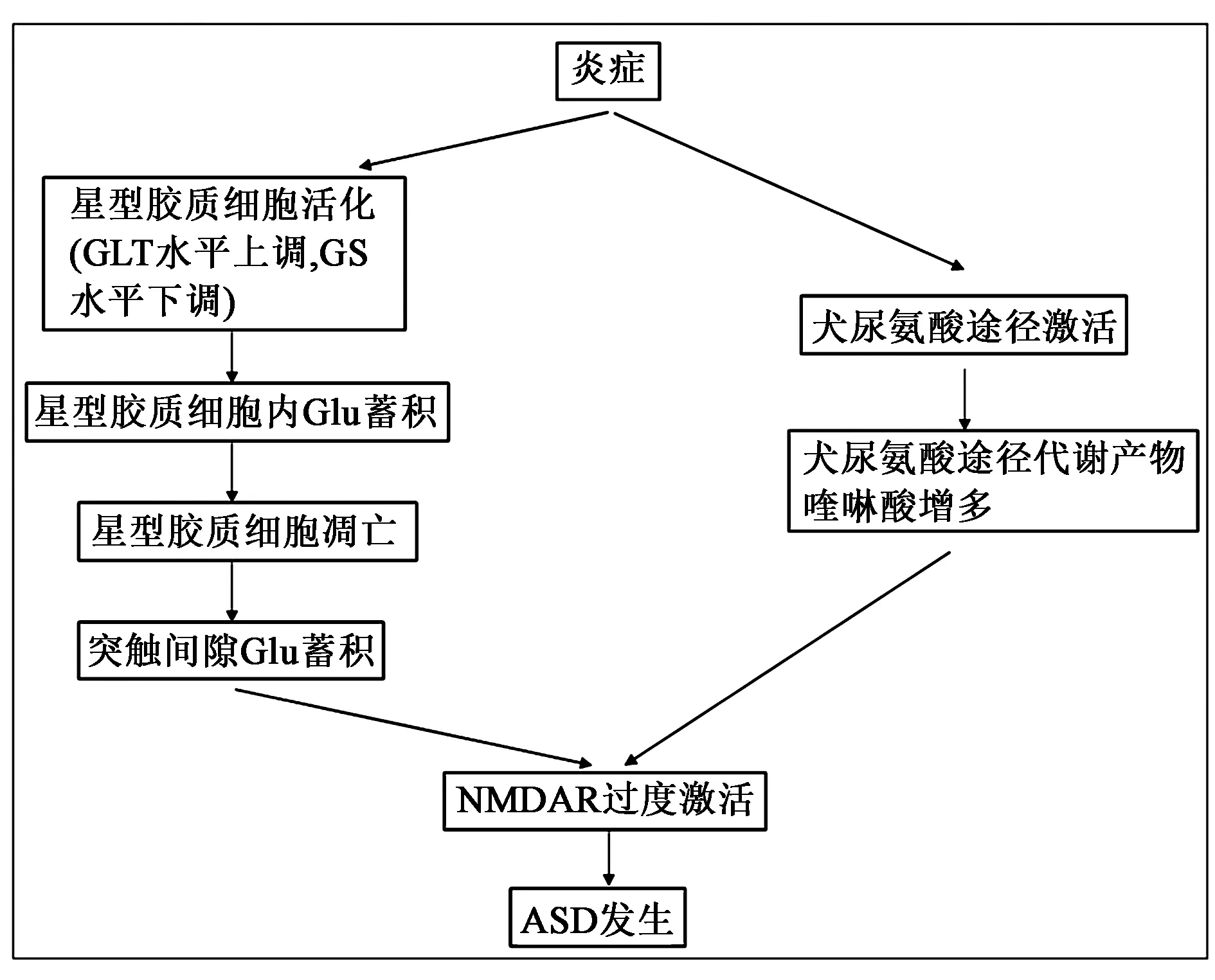
图2 炎症通过影响谷氨酸-谷氨酰胺循环导致ASD发生Fig.2 Inflammation leads to ASD by affecting glutamate-glutamine cycle
3 自身免疫性抗体与ASD
自身免疫性抗体由B细胞合成,CD19是B细胞表面重要膜抗原。对ASD患者外周血流式细胞分型,可发现CD19阳性细胞计数较正常人群高[32]。部分ASD患者发病与自身免疫性抗体相关,例如:1型糖尿病绝大多数为自身免疫性疾病,母亲患1型糖尿病,其子女ASD风险也更高[33]。但Connery等[34]对82名ASD患儿自身免疫性脑炎相关的多种抗体进行检测,发现NMDAR血清抗体均阴性,仅有5%患儿GAD65抗体阳性。当然,抗NMDAR抗体脑炎患儿可表现为ASD样症状[35],说明该抗体可能与ASD发病相关,但并不是由于抗体直接影响谷氨酸-谷氨酰胺循环所致,因为NMDAR抗体阳性的ASD患儿突触上NMDAR并未被破环[36]。自身免疫抗体可能是在大脑形成免疫复合物介导炎症,从而间接导致该循环异常[37]。
4 基因异常与ASD
多种ASD相关基因与谷氨酸-谷氨酰胺循环异常相关,且ASD严重程度与该循环存在遗传关联[38]。NMDAR基因表达异常会导致ASD发生,其离子通道开放需同时激活突触前膜和突触后膜,这种独特开放机制使NMDAR对于突触可塑性具有重要影响。长时程抑制 (long-term depression, LTD)和长时程增强 (long-term potentiation, LTP)是突触可塑性的两种关键形式。
NMDAR轻度去极化时,Ca2+少量进入突触后神经元,突触后膜AMPAR被移除且去磷酸化,LTD发生。Neuroligin基因家族编码突触后细胞黏附分子,对突触的发育和成熟有重要作用。Neuroligin1是该家族成员之一,Neuroligin1基因缺失杂合子小鼠海马区NMDAR介导的LTD减弱,并表现为ASD样症状[39]。同样,该家族另一基因,R451C-neuroligin3发生突变,也会减弱LTD,导致ASD发生[40]。但Lee等[41]提到,ASD相关基因突变可能是减弱mGluR介导的LTD,而非NMDAR介导的LTD。
LTP是突触可塑性的另一种形式。当NMDAR被激活,Ca2+大量进入突触后神经元,非突触AMPAR插入突触后膜且AMPAR被磷酸化,LTP发生。ASD相关基因突变影响NMDAR介导的LTP也可能是疾病发生的一种机制。Rac1是一种小G蛋白,具有GTP酶活性,当其被鸟嘌呤核苷酸交换因子(guanine nucleotide exchange factor,GEF)激活,与GTP结合时,便能促进肌动蛋白聚合。Dock4属于GEF成员,由DOCK4基因编码,可激活Rac1,DOCK4基因敲除小鼠表现为ASD样行为,且海马CA1区NMDAR密度减少,NMDAR介导的LTP也被抑制,而药物激活NMDAR可减轻这些小鼠的社会认知障碍[42]。总之,ASD相关基因表达异常可能影响谷氨酸-谷氨酰胺循环,并进一步影响突触可塑性,从而导致疾病的发生。
5 展望
ASD临床表现不尽相同,病因也是复杂多样,导致该疾病诊断和治疗难度颇大,探究众多病因的内在联系,明确ASD发病核心机制,对于疾病的研究显得极其重要。谷氨酸-谷氨酰胺循环异常可能是ASD的核心发病机制,炎症、自身免疫性抗体和ASD相关基因异常等经典病因均可能通过影响该循环,导致疾病的发生。当前,谷氨酸-谷氨酰胺循环异常具体如何导致ASD发生,相关研究结论并不一致。后续临床研究需要尽可能扩大样本量,且研究对象尽可能同质。本文为简化ASD病因及分型提供了一种新思路,即ASD发病相关病因较多,但它们均可能是通过影响谷氨酸-谷氨酰胺循环导致疾病的发生,那么,未来或许可针对该循环这一共同靶点对ASD进行有效治疗。
猜你喜欢
传染病信息(2022年2期)2022-07-15
昆明医科大学学报(2021年3期)2021-07-22
食品与生物技术学报(2021年4期)2021-01-17
岭南现代临床外科(2020年5期)2020-12-13
初中生世界·七年级(2018年7期)2018-09-07
中成药(2017年6期)2017-06-13
中国洗涤用品工业(2017年2期)2017-04-16
中国调味品(2017年2期)2017-03-20
中国比较医学杂志(2017年5期)2017-01-17
中国药理学与毒理学杂志(2015年3期)2015-12-16
