
汉坦病毒感染致血小板减少的机制
2021-03-25 08:10王晓艳申焕君王平忠
传染病信息 2021年1期
王晓艳,杜 虹,李 璟,姜 泓,申焕君,王平忠
汉坦病毒是一个“属”的名称,归布尼亚病毒科。该“属”病毒包括引起肾综合征出血热(hemorrhagic fever with renal syndrome,HFRS)的汉滩病毒(hantaanvirus,HTNV)、汉城病毒、普马拉病毒、多布拉伐病毒、泰国病毒,引起汉坦病毒心肺综合征的辛诺柏病毒、纽约病毒、安第斯病毒,以及与人类关系尚不清楚的一组病毒,如希望山病毒、图拉病毒等。其中,HTNV和汉城病毒分别是我国姬鼠型(Ⅰ型)和家鼠型(Ⅱ型)疫区引起HFRS的主要流行毒株。HFRS流行于欧亚大陆,而汉坦病毒肺综合征流行于北美洲和南美洲。全球每年约有6万~10万HFRS患者,90%以上分布在中国,病死率可达5%~10%[1]。尽管国内外学者对汉坦病毒及其相关疾病的研究已取得了重要进展,但发病机制尚未完全阐明,也无特效治疗药物。汉坦病毒主要感染人血管内皮细胞,引起毛细血管广泛损伤,通透性增加。临床上HFRS主要表现为发热、低血压休克、出血和肾功能衰竭。毛细血管通透性增加是上述临床表现的病理基础,与HFRS患者出现严重血小板减少密切相关。近年,虽然汉坦病毒感染致血小板减少的机制尚不完全明确,但也取得了一些重要进展。
1 血小板的生成及调控
血小板由多能造血干细胞(pluripotential hematopoietic stem cell,PHSC)定向分化而成。从PHSC形成血小板主要经历了以下阶段:PHSC定向分化为巨核细胞(megakaryocyte,MK)、MK增殖成熟、前血小板形成和血小板释放。在此过程中,细胞表面标志物和核内染色体均发生了显著变化。CD61(整合素β3)、CD41(整合素αIIb)和CD42(GPIb复合体)是MK分化的标志物。CD41和CD61构成异二聚体的受体复合体GPIIb/IIIa(αIIbβ3整合素)存在于MK谱系细胞(从MK祖细胞到血小板)的表面。CD41表达水平随细胞的成熟而增加,而CD42(GPIb复合体:GPIbα、GPIbβ、GPⅨ和GPⅤ)表达稍晚于CD41,它对应晚期分化阶段。当启动血小板蛋白合成后,CD34消失。与此同时,MK前体通过核内有丝分裂增加染色体倍数。MK成熟后,具有伪足的前血小板断裂脱落生成血小板。
MK分化和血小板释放过程受特异性转录因子NF-E2和GATA-1的调控。NF-E2是由造血特异性亚基(p45)和广泛表达的亚基(p18)构成的异二聚体。NF-E2在MK成熟、前血小板形成和血小板释放以及建立血小板正常功能中起非常重要的作用[2]。目前,已鉴定的NF-E2靶基因主要有:β微管蛋白(表达于MK分化的晚期,是形成前血小板所需微管的主要成分)、Rab27b(一种小的G蛋白,与血小板颗粒运输有关,在前血小板形成中起重要作用)、半胱天冬酶-12(高表达于成熟的MK,但在血小板中无表达)和凝血烷合成酶(该酶可促进血小板凝集)[3]。GATA-1是GATA家族成员之一。GATA-1高表达于红细胞、MK和肥大细胞。GATA-1缺陷表现为明显的血小板减少和严重贫血[4]。依赖GATA-1表达的基因主要是红系和MK谱系的特异性基因,包括红细胞生成素受体、α/β球蛋白、c-mpl[血小板生成素(thermoplastic polyolefin,TPO)的受体]、血小板糖蛋白 [Ibα、Ibβ、IIb(αIIb)、IX、VI]和血小板因子-4(PF-4/CXCL-4)等[5]。可见,NF-E2和GATA-1通过调控血小板生成过程所必需的蛋白,调节血小板的形成。
此外,血小板生成过程还受一些细胞因子调节。转化生长因子 β1(transforming growth factor-β1,TGF-β1)、IFN-α和CXC趋化因子(如IL-8、IP-10、PF-4、β-TG等)是MK生成的负向调节因子,其中最重要的是TGF-β1。TGF-β1通过自分泌或旁分泌机制在造血系统的多个阶段均发挥多效调节作用[6]。TPO(也称MK生长和发育因子)、IL-3、粒细胞-巨噬细胞集落刺激因子和干细胞因子可促进MK增殖,TPO、IL-6、IL-11和白血病抑制因子能促进MK多倍体化和细胞质成熟。此外,研究还发现,增殖诱导配体APRIL(属于TNF超家族的细胞因子)也是MK生长和分化过程关键的因子,其受抑制可减少成熟MK数量[7]。
由此可见,转录因子和细胞因子在血小板生成调控中发挥着重要作用。转录因子是处于“中心”地位的“内源”因素,通过调控血小板生成过程中所必需的蛋白,直接影响血小板的形成,而细胞因子则是一种“外源”因素,为血小板形成提供必需的“微环境”,在生理条件下,二者协同以保证血小板的正常生成,并维持其正常数量。但在疾病条件下(如病毒感染、药物治疗等),可能抑制转录因子的表达或细胞核移位,或者使细胞因子表达异常(如促进血小板生成的因子下降,而抑制血小板生成的因子上升),最终影响干细胞分化和MK成熟,导致血小板生成障碍,引起血小板减少。
2 病毒感染致血小板减少的机制
多种病毒感染均可致血小板减少,其机制包括“中心机制”和“外周机制”(图1)。
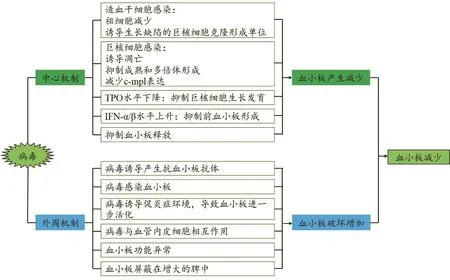
图1 病毒感染致血小板减少的机制(根据文献总结)Figure 1 Mechanism of thrombocytopenia induced by viral infection (based on literature)
2.1 中心机制 血小板生成过程的任一环节异常,均可致血小板数量减少。HIV-1 gp120 V3环序列可抑制CD34+祖细胞向MK谱系的特异性分化,对MK生成的抑制可能通过c-mpl的表达减少及其随后的信号传导蛋白和转录激活物-5活化下降而实现[8]。人疱疹病毒-6感染CD34+细胞可抑制集落形成单位MK的形成[9]。人巨细胞病毒感染CD34+造血干细胞,可抑制集落形成单位MK的增殖和分化[10],人巨细胞病毒感染MK系(CHRF-288-11),可诱导凋亡,致血小板减少[11]。
病毒性出血热是由沙粒病毒科、布尼亚病毒科、纤丝病毒科和黄病毒科的病毒引起的一种急性全身性发热综合征。引起病毒性出血热的病原体虽有不同,但其标志特征都是血管渗漏、血小板减少、凝血功能不全和出血[12]。Junin病毒(Junin virus,JUNV)是沙粒病毒科的成员,感染可引起阿根廷出血热。用JUNV感染TPO体外诱导的脐血CD34+细胞。结果表明,JUNV感染不影响CD34+细胞的增殖、存活和定向分化,但可降低前血小板形成和血小板释放。其原因主要是JUNV感染引起转录因子NF-E2的表达水平下降,而GATA-1的表达水平基本没有变化[13]。登革病毒(dengue virus,DV)属黄病毒科成员,感染可引起登革热、登革出血热和登革休克综合征。在骨髓中,DV抑制MK的发育是其诱导血小板减少的关键机制[14-15]。
因此,病毒感染可能调控造血干细胞分化、MK增殖成熟、前血小板形成和血小板释放,最终影响血小板生成。
2.2 外周机制 除上述“生成障碍”(中心机制)外,“外周破坏”(外周机制)也是一个重要因素。这些外周机制主要包括:血小板抗体介导的活化和清除、血小板与内皮细胞的相互作用、血小板功能异常、病毒感染血小板和弥漫性血管内凝血(disseminated intravascular coagulation,DIC)。在DV感染者中,上述外周机制的一些因素参与了血小板减少症的发生[16]。HCV感染可引起不同程度的血小板减少。研究发现,在严重血小板减少组(<100 000/μl),骨髓抑制和自身免疫(血小板抗体)破坏共存。在中等程度血小板减少组(100 000~125 000/μl),26.9% 的患者仅有骨髓抑制,11.5%的患者仅有自身免疫破坏,61.5%的患者两种机制共存。在轻度血小板减少组(126 000~149 000/μl),其机制主要是自身免疫破坏[17]。HBV、HCV相关性肝硬化患者,血浆TPO水平下降,血小板更新加快,血小板产生减少[18],表明该类患者血小板减少是一个多因素结果,既有血小板清除增加的因素,也有血小板生成减少的因素。
由此可见,不同病毒引起血小板减少的机制可能有别。在正常生理条件下,血小板数量处于恒定水平,但在疾病条件下(如病毒感染、药物治疗等),可能引起血小板生成减少和/或外周破坏增加,最终导致血小板数量减少。血小板减少症患者出血的风险随着血小板数量和功能的下降而增加。因此,了解病毒感染致血小板减少的机制,对临床治疗策略和发病机制研究及药物设计具有重要的指导意义。
3 汉坦病毒感染致血小板减少的机制
尽管许多病毒感染可致血小板减少,但汉坦病毒感染引起的急性血小板减少具有更明显的临床表现和更严重的后果(如出血、低血压休克、肾功能衰竭等)。对汉坦病毒心肺综合征患者的研究发现,轻型患者入院时血小板计数越低越易进展至重型[19]。临床上,HFRS患者在低血压休克期,血小板数量急剧减少,直至少尿期血小板仍处于较低水平,并出现严重的血管渗漏,是HFRS患者的主要死因之一[20]。出血的风险随着血小板数量减少和功能的下降而增加,表明HFRS患者出现的血管渗漏与病程中血小板减少有密切的关系。
汉坦病毒感染致血小板减少的机制尚不完全清楚,但近年也取得了一些重要进展。血小板由造血干细胞分化形成的MK产生,在维持血小板数量平衡中起至关重要的作用。HFRS患者病程中出现的严重“血小板减少”,不仅与“外周破坏增加”有关,而且还可能与“血小板生成障碍”有关。
目前关于外周破坏机制的研究包括:①αIIbβ3整合素[即血小板糖蛋白IIb/IIIa,其中β3亚基(CD61)是致病汉坦病毒的主要受体,也是MK分化的早期标志]是血小板表面最丰富的受体,在血小板活化、聚集和粘附中起重要作用[21]。研究发现,HTNV和HTNV感染的内皮细胞均可结合静止的血小板,阻止其活化[22],提示HTNV感染不仅减少了血液中游离血小板的有效浓度,而且还可能导致血小板功能异常。②HFRS患者血小板CD61表达水平明显高于健康对照,其表达越高,血小板减少症的发生率就越高。同时,CD61表达水平在少尿期与血小板数量呈负相关[23],提示汉坦病毒感染可上调CD61表达,其结果是增加了病毒感染,但影响了血小板的数量。③普马拉病毒感染可引起流行性肾病 (流行于欧洲的一种轻型HFRS),研究发现在急性流行性肾病病程中,血小板减少与自然抗凝血降低、凝血酶时间缩短及纤维蛋白溶解增加有关[24],提示DIC参与了血小板减少症的发生,因DIC须消耗大量血小板。④在HFRS患者和流行性肾病患者中发现,血小板糖蛋白Ia、IIb/IIIa具有多态性,这种多态性可能改变了分子构象,导致血小板功能异常,引起血小板减少[25-26]。
MK生成是血小板形成过程最重要的阶段。与“血小板生成障碍”相关的研究包括:用HTNV感染MK系(HEL)和人原代MK(CD34+细胞经TPO体外诱导5 d产生于培养液中的较大细胞)可上调β3整合素(CD61)表达,而CD42(MK生成的晚期标志)表达没有变化[27],提示HTNV感染可能抑制MK的成熟。本课题组用HTNV感染MK系(HEL)12 h、24 h、36 h,RT-PCR检测NF-E2、GATA-1和c-mpl mRNA的表达。结果发现,感染36 h后,与未感染组相比,NF-E2和GATA-1表达水平均有下降,而c-mpl表达水平基本没有变化(结果待发表),提示HTNV感染可能抑制MK转录因子的表达,从而减少血小板的产生。其次,用ELISA测定了HFRS患者不同病期TPO和TGF-β1水平,发现从发热期到恢复期TPO含量虽有下降,但无明显差异,其受体(c-mpl)的表达也无明显变化。而低血压休克期TGF-β1含量显著高于其他各病期,TGF-β1含量越高,血小板数量越少(结果待发表),提示TGF-β1是血小板生成负向调控的主要因子,在HFRS患者血小板减少的形成中可能起重要作用。可见,汉坦病毒感染引起的血小板减少,“中心机制”和“外周机制”可能都参与其中,最终导致血小板减少。
4 展 望
尽管血小板减少是汉坦病毒相关疾病的重要特征,但汉坦病毒感染致血小板减少的机制迄今尚不完全清楚。血小板减少可由生成障碍和/或外周破坏引起。前者因血小板生成“受阻”所致(中心机制),后者主要包括免疫介导的血小板活化和清除、血小板与内皮细胞的相互作用、血小板功能异常、DIC和病毒感染血小板(外周机制)等。近年,关于汉坦病毒感染致血小板减少的报道,大都集中在外周机制方面,而对于中心机制的研究很少。未来系统阐明汉坦病毒感染致血小板减少的原因,将为发病机理和治疗策略研究及药物设计提供重要的科学依据。
猜你喜欢
中老年保健(2022年1期)2022-08-17
清华金融评论(2022年4期)2022-04-13
中国民间疗法(2021年5期)2021-06-09
国际放射医学核医学杂志(2021年10期)2021-02-28
房地产导刊(2020年7期)2020-08-24
兽医导刊(2019年1期)2019-02-21
猪业科学(2018年8期)2018-09-28
河北医学(2016年5期)2016-12-01
癌症进展(2016年10期)2016-03-20
中国药理学通报(2014年2期)2014-05-09
