
CRISPR基因编辑技术在微生物合成生物学领域的研究进展
2021-03-18 07:52李洋申晓林孙新晓袁其朋闫亚军王佳
合成生物学 2021年1期
李洋,申晓林,孙新晓,袁其朋,闫亚军,王佳
(1北京化工大学化工资源有效利用国家重点实验室,北京 100029,中国;2美国佐治亚大学工程学院,佐治亚州,阿森斯 30602)
进入新世纪以来,随着科学与技术的蓬勃发展,出现了很多交叉学科。合成生物学是其中一门重要的新型交叉学科,主要涉及基因工程、系统生物学、生物化学、生物信息学、计算机科学和工程学等领域。微生物合成生物学作为合成生物学的一个重要分支,是以微生物为载体,通过改造已有微生物细胞或设计并创建新的微生物元件,使底盘生物实现其特定的生物学功能。在改造或创制这些微生物的过程中,需要对底盘生物基因组进行精简、插入或重构,而高效精准的基因编辑技术成为解决这些问题的有效手段,为微生物合成生物学的进一步发展提供了强有力的支持。
基因编辑技术作为基因工程、代谢工程、医学研究等领域的重要技术,一直以来都是研究热点。传统的基因编辑方法如同源重组,存在打靶效率低、操作时间长和操作烦琐等问题。为了解决这些问题,陆续出现了P1 转导技术[1]、锌指核酶技术(zinc-finger nucleases,ZFNs)[2]、RNA 干扰技术(RNAi)[3]和转录激活因子效应蛋白核酸酶技术(transcription activator-like effector nucleases,TALENs)[4]等。近年来,研究者们又发明了CRISPR (clustered regularly interspaced short palindromic repeats)技术[5],相较于上述基因编辑技术,CRISPR 技术具有操作难度小、编辑成本低、打靶效率高、无痕编辑等优点,在微生物合成生物学领域被广泛应用和研究,并由此开发和设计出了大量新的基因编辑元件、工具和基因线路[6]。
基于此,国内外有大量研究人员对CRISPR 基因编辑技术从发现历史、分类[7]、作用机理[8]及在微生物基因改造领域中的应用[5,6,9,10]等方面进行了综述。近几年来,随着合成生物学和代谢工程的迅猛发展,利用CRISPR 基因编辑技术创建高效的微生物细胞工厂生产特定化学品、燃料和药品不断取得新的突破。本文综述了CRISPR 基因编辑技术在不同微生物合成生物学中的应用,重点介绍了应用不同技术在不同微生物中生产特定产品的最新进展,阐述了在CRISPR/Cas9 系统基础上发展的CRISPR/Cas12a、CRISPR/Cas13 等技术的研究现状,阐明了该技术在微生物合成生物学领域的重要性,总结并展望了该技术在微生物合成生物学领域未来的发展方向。
1 CRISPR技术发展及分类
CRISPR 技术的高速发展,归功于无意中发现的一种未知片段。1987 年,Ishino 等[11]在大肠杆菌中发现了一段重复了5次,长度为29 bp的序列,每段重复序列被长度为32 bp 的杂乱序列隔开。2000 年,西班牙研究者Mojica 等[12]通过收集不同微生物中这种类型的重复序列,发现这种序列在微生物中广泛存在。直到2002 年,Jansen 等[13]观察了大量细菌和古细菌基因组后,创造了缩写CRISPR 代表之前提到的重复序列。之后,Barrangou 等[14]证实了CRISPR 序列被用于微生物对噬菌体免疫的猜想,揭示了CRISPR 序列在微生物中的主要功能。2008 年,Brouns 等和Marraffini等[15-16]对CRISPR/Cas 系统的研究揭示了CRISPR转录并加工得到的crRNA(CRISPR-RNA)对DNA 具有靶向作用,因此推测CRISPR 系统具有作为基因编辑工具的潜力。2011年,Charpentier课题组发现RNA 酶Ⅲ参与crRNA 成熟前的修饰[17],次年与Doudna 课题组合作完成单链嵌合RNA(single chimeric RNA)的研究[18]。2013 年,张锋团队实现了CRISPR/Cas9 系统在哺乳动物细胞中进行多基因编辑的功能[19],将CRISPR技术的应用扩大化。目前为止,CRISPR 技术在微生物混合筛选方面也有相关的应用[20-23]。随着CRISPR 系统研究得越来越深入,研究者们实现了该系统在小鼠[24]、斑马鱼[25]、线虫[26]、拟南芥[27]、真菌[28]和细菌[29]等多种生物基因编辑中的应用。
CRISPR/Cas 系统由识别组件CRISPR 和切割组件Cas(CRISPR-associated proteins)组成。其中,识别组件CRISPR序列经过转录形成crRNA前体(pre-crRNA),随后pre-crRNA 经过加工形成成熟的具有靶向识别功能的crRNA;切割组件Cas是一种核酸内切酶,可与成熟的crRNA 形成RNA-蛋白质结合体,随着crRNA 与特定的DNA 序列特异性识别结合,Cas 切断靶向的DNA 链,使基因产生缺口(double-strand breaks,DSBs),实现“打靶”;最后通过同源重组(homology-directed repair,HDR)[30]或非同源末端连接(nonhomologous end joining,NHEJ)[31]等方式进行基因修复,达到基因编辑的目的。
根据Cas蛋白的结构和进化关系,CRISPR/Cas系统起初被分成3 种类型:Ⅰ型、Ⅱ型和Ⅲ型[32]。其中Ⅰ型和Ⅲ型系统的Cas蛋白均是多亚基蛋白质;而Ⅱ型系统的Cas蛋白则是单亚基蛋白质。随着研究的深入,更多类型的CRISPR/Cas 系统被发现,如Ⅳ型、Ⅴ型和Ⅵ型,其中Ⅳ型是多亚基Cas 蛋白,Ⅴ型和Ⅵ型为单亚基Cas蛋白[33]。另外,根据Cas 蛋白功能不同,可将CRISPR/Cas 系统分为两类:一类是由单亚基蛋白完成定点靶向和切割过程,目前该类系统仅在细菌中发现;另一类则是多亚基结构的crRNA-蛋白复合体完成crRNA 加工、靶向定位和剪切[7],这类系统广泛存在于古细菌及细菌中。但是多亚基的CRISPR/Cas系统较为复杂,因此在基因编辑上应用并不多,而单亚基蛋白如Cas9 蛋白[34]、Cas12a 蛋白[35]等,由于其原理简单、操作方便等优点,应用较为广泛。因此,本文将重点介绍CRISPR/Cas9在微生物合成生物学中的应用及研究进展。
2 CRISPR/Cas9 在微生物合成生物学中的应用
Cas9 蛋白是一种单亚基的核酸酶,是目前微生物合成生物学研究中使用的CRISPR 系统中最常用的切割组件,通常将该类系统称为CRISPR/Cas9系统。该系统在进行基因编辑时,tracrRNA(trans-activating crRNA)、pre-crRNA 和Cas9 蛋白先被转录和表达出来;接着,tracrRNA 启动RNA酶Ⅲ修饰pre-crRNA,形成成熟的crRNA;然后crRNA、tracrRNA 和Cas9 蛋白形成复合物,通过crRNA 的识别作用将复合体靶向目标DNA,与此同时,tracrRNA 激活Cas9 蛋白;接着,Cas9 发挥核酸酶的作用切割目标DNA,使其双链断裂产生DSBs[17]。产生DSBs 后,细胞可通过不同修复方式修复DNA 双链(图1)。在该系统中,crRNA 和tracrRNA 可被一种单链的向导RNA(single-guide RNA,sgRNA)取代,简化识别组件RNA。为了确保复合体的成功识别,在sgRNA 的靶序列下游必须含有PAM序列(protospacer adjacent motif)[18]。PAM 是位于靶向DNA 的3′端后长度为3 bp 的序列,供sgRNA 和Cas9 蛋白复合体识别,其序列组合取决于Cas9 蛋白种类,通常为NGG[36]。CRISPR/Cas9系统通过上述过程可在微生物中实现基因的缺失、增添和替换等基因编辑操作,使微生物表现出与编辑前不同的特性,实现特定的生物学功能。微生物合成生物学的两个最重要的目的分别是改造已有的微生物底盘细胞或创造新的底盘细胞,使其实现特定的生物功能。因此,设计和构建元件库来改造、编辑或合成微生物底盘细胞基因组是合成生物学的重点研究内容。CRISPR 系统的开发与发展为微生物合成生物学的研究提供了一个非常简便易操作的基因编辑技术,因此,本文将重点回顾与总结该技术在微生物合成生物学方面的研究与应用。
2.1 CRISPR-Cas9技术在大肠杆菌基因编辑中的应用进展
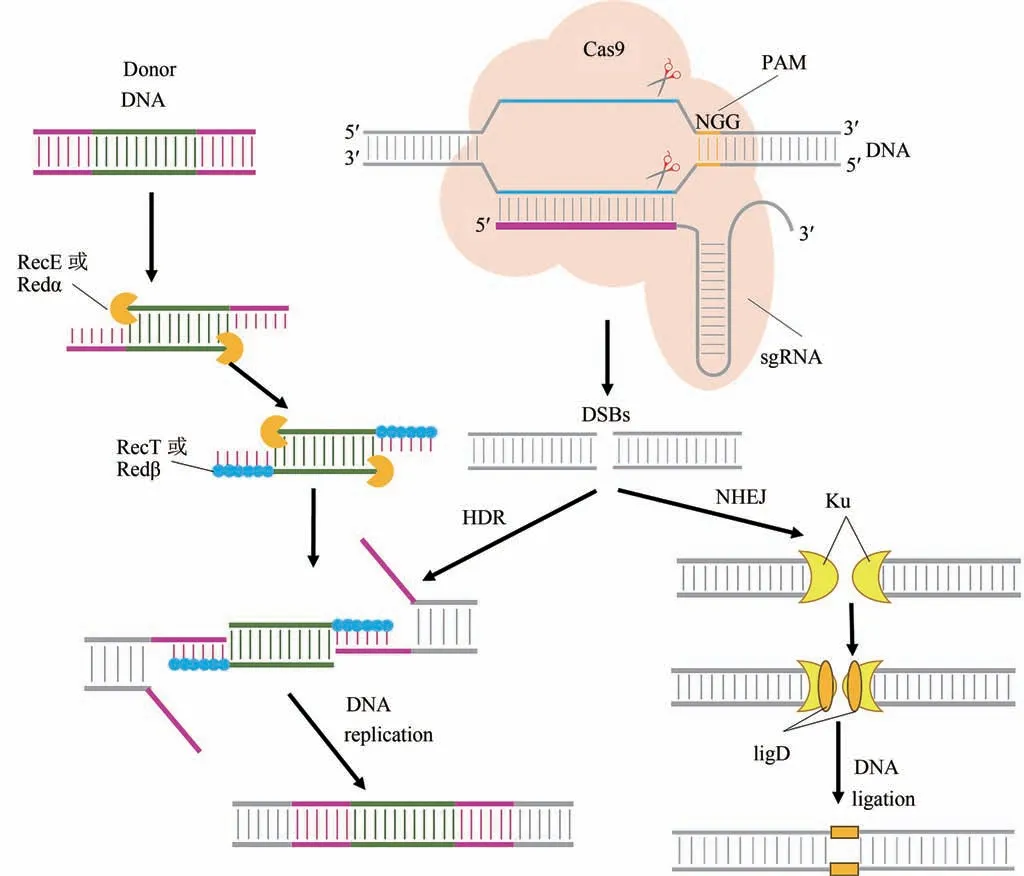
图1 CRISPR/Cas9系统切割机制及修复机制图[Cas9蛋白中蓝色代表识别DNA被识别的位点,橙色代表PAM序列,深紫色代表sgRNA上的识别序列;在HDR过程中,浅紫色代表同源序列,绿色代表进行替换的序列,橙色蛋白为RecE或Redα(负责供体DNA单链切割),蓝色蛋白为RecT或Redβ(负责黏性末端的保护以及和基因断裂位点的结合);在NHEJ过程中,黄色蛋白表示Ku(负责保护断裂位点两侧),橙色蛋白为ligD(负责连接断裂位点)]Fig.1 The cleavage and repair mechanism of CRISPR/Cas9 systems(Blue represents the recognized site of DNA,orange represents PAM sequence,and dark purple represents the recognition sequence by sgRNA.During HDR,light purple represents homologous sequences,green represents substituted sequences,orange represents RecE or Red responsible for donor DNA single strand cutting,and blue represents RecT or Red responsible for protecting sticky ends and binding to the breaking sites of genes.During NHEJ,the yellow protein represents Ku responsible for protecting the fracture site,while the orange protein is ligD responsible for connecting to the fracture site)
大肠杆菌(Escherichia coli)由于其易于培养、增殖时间短、对环境的耐受性强、基因操作容易等特点,是微生物合成生物学研究中重要的模式生物[37]。因此,在大肠杆菌中构建CRISPR/Cas9技术平台十分重要。2013年,Jiang等[38]首先在大肠杆菌使用了CRISPR/Cas9 系统,通过Cas9 切断目的基因,由λ-Red 蛋白修复实现目的基因的编辑,将突变序列引入宿主,证实了该系统在大肠杆菌中可有效进行基因编辑,突变率达到65%。2015 年,天津大学赵学明团队改造了CRISPR/Cas9 系统,使其可进行迭代基因编辑,从而降低多基因编辑的循环时间,该系统同时对3个基因进行编辑的效率接近100%[39]。同年,中国科学院上海生物科学研究所杨晟团队成功构建了一个大肠杆菌CRISPR/Cas9基因编辑平台,通过双质粒系统巧妙地同时实现了基因的断裂、重组修复和质粒的消除[40]。该团队简化了编辑方法,只需要替换质粒中与靶DNA识别的20 个碱基(N20),即可编辑不同的基因,而在质粒上引入多个具有特定N20结构的sgRNA 的序列即可完成多基因的编辑[40]。2016 年,Zhao等[41]通过一个质粒就实现单一基因的编辑,节省了质粒构建的时间。编辑平台的建立在一定程度上方便了CRISPR/Cas9 技术在大肠杆菌中的应用,同时提高了编辑效率,节约了时间。但有些问题被暴露出来,如供体基因(donor DNA)长度不能太长、一次编辑的基因数量不能过多等。2016 年,为了将多个基因或大片段基因整合进大肠杆菌,Chung 等[42]通过优化实验条件并结合λ-Red 重组蛋白和线性dsDNA,实现供体DNA 和lacZ基因的高保真替换,效率达99%,同时实现了长度分别为2.4 kb、3.9 kb、5.4 kb 和7.0 kb DNA 片段的整合,效率分别达到91%、92%、71%和61%。研究发现,随着引入大肠杆菌基因组片段长度的增加,编辑效率逐步降低,为了进一步提高转入长片段的基因编辑效率,2018 年,Li 等[43]采用将大片段分割成多个片段整合的方法,每轮整合只提供小同源片段,同时优化了gRNA(guide RNA)和Cas9 蛋白,成功将长度为15 kb 的大片段引入大肠杆菌基因组。在大肠杆菌基因编辑的过程中,通常需要引入HDR 系统如Red 重组系统完成对于编辑后DSB的修复,而NHEJ修复很难在原核生物中实现[44]。为了使大肠杆菌摆脱对HDR 系统的依赖,提高修复效率,有研究者将真核生物的NHEJ修复系统引入大肠杆菌。Zheng等[45]通过引入耻垢分枝杆菌(Mycobacterium smegmatis)的NHEJ 系统,在大肠杆菌中突变了lacZ基因,并敲除了glnALG操纵子和两段长度分别为67 kb和123 kb的大片段DNA。为了进一步解决大肠杆菌对外源HDR 系统或NHEJ 系统的依赖,Huang 等[46]通过优化大肠杆菌天然的末端连接(ENEJ)系统,实现了83 kb以下DNA片段的缺失或失活。这些研究都有效地提高了大肠杆菌基因编辑的效率,也为CRISPR/Cas9编辑系统在大肠杆菌中的应用提供了有力保障。
微生物合成生物学一个重要目标是对已知的生命体进行基因编辑,高效合成目标产品[47]。因此,有大量的CRISPR/Cas9 系统被应用于提高目标产物生产效率的研究。天津大学研究团队利用CRISPR/Cas9 基因编辑平台强化EMP 途径和β-胡萝卜素生产相关途径,构建了超过100种基因突变的突变文库,最终,含有15 个修饰基因的大肠杆菌在发酵罐中生产了2.0 g/Lβ-胡萝卜素[39]。夏军等[48]利用杨晟团队构建的CRISPR/Cas9 基因编辑平台通过敲除丙酮酸羧化酶基因(ppc)、脂酰辅酶A合成酶基因(fadD)等基因,使脂肪酸产量增加了3.7%。Li 等[43]通过CRISPR/Cas9 技术将pyr操纵子、ppc和核糖磷酸焦磷酸激酶基因(prs)整合到大肠杆菌染色体中,在摇瓶中生产了5.6 g/L 尿苷。Abdelaal 等[49]敲除木糖产乙醇菌株的内源性乙醇生产基因后,引入生产正丁醇的相关基因,在大肠杆菌中生产了4.32 g/L 正丁醇。Jung 等[50]在大肠杆菌中利用CRISPR/Cas9 技术敲除了丙酮酸甲酸裂解酶(pflb)、乳酸脱氢酶(ldhA)、乙醇脱氢酶(adhE)和转录调节因子(fnr)的4 个编码基因,降低副产物的生成,并过表达转氢酶基因pntAB使聚羟基脂肪酸酯在摇瓶中产量达到32 g/L。Zhao 等[51]在原有研究基础上用CRISPR/Cas9 技术敲除了琥珀酰辅酶A 连接酶基因(sucD),促进了己二酸合成前体琥珀酰辅酶A 的积累,使目标产物己二酸的产量达到68.0 g/L。这些研究表明,利用CRISPR/Cas9 系统对大肠杆菌进行基因编辑的技术已十分成熟。但是,大肠杆菌仍具有不耐受强酸、有机溶剂和噬菌体等缺点。因此,提高大肠杆菌对目标产物或噬菌体的耐受性可在一定程度上提高目的产物的产量或降低噬菌体污染风险。Seo 等[52]用CRISPR/Cas9 技术在大肠杆菌基因组中插入了非编码RNA 基因(dsrA)和DNA 结合转录激活因子RcsB 基因(rcsB),提高了大肠杆菌对庚酸的耐受性,构建了适于生产庚酸的工程大肠杆菌。Ou 等[53]通过引入N&P 系统在宿主中表达甲酰胺酶和亚磷酸酯脱氢酶使目标大肠杆菌可以以甲酰胺和亚磷酸酯为氮源和磷源生长,同时引入CRISPR/Cas9 系统协助大肠杆菌增强抵抗T7 噬菌体攻击的能力,开发出了耐受性较强的大肠杆菌BL21(DE3)菌株,在工业发酵中具有很大的应用潜力。综上所述,CRISPR/Cas9技术在大肠杆菌合成生物学中的应用非常广泛且较为成熟,但是该系统在细菌中的应用还存在脱靶率较高、编辑成功率较低、多位点同时编辑效率低且应用困难、PAM 序列依赖等问题,因此需要通过进一步的研究和开发如拓展PAM 序列、计算机辅助设计蛋白定点突变提高切割效率等,来完善和提高该技术在大肠杆菌基因编辑中的大规模应用。
2.2 CRISPR-Cas9 技术在酵母基因编辑中的应用进展
酿酒酵母(Saccharomyces cerevisiae)是常用真核模式生物,不仅可以生产酒精[54],还常常作为合成生物学和代谢工程等领域的宿主菌,生产多种高附加值化学品[55],如青蒿素[56]等。为了满足科学研究和工业化的需求,CRISPR/Cas9 系统在酵母菌合成生物学中的研究和应用越来越多,也日趋成熟。在sgRNA 的设计方面,已经出现了一些非常方便的工具网站,如“Yeastriction web tool”,这些工具的出现极大地方便了在酿酒酵母中进行基因编辑的操作[57]。早在2013 年,就有研究者使用90 bp 的双链DNA 作为同源修复的模板,在can1位点实现100%的编辑效率[58]。2015 年,Horwitz 等[59]证明了线性化质粒与多个sgRNA 和供体DNA 的共转化促进了大片段高效多重基因整合,并将6 个总长度为24 kb 的片段成功整合在酵母基因组中。但是,这些供体DNA同时整合的概率较低,因此有研究人员将HDR 系统引入到sgRNA 上,构建了同源整合CRISPR(homology-integrated CRISPR,HI-CRISPR)系统。利用此方法,敲除了精氨酸透性酶基因(can1)、磷酸核糖酰氨基咪唑羧化酶基因(ade2)、赖氨酸透性酶基因(lyp1)三个基因[60]。Ryan 等[61]将δ 肝炎病毒核糖酶与sgRNA相连以提升sgRNA 丰度,构建多重CRISPR(CRISPRm)系统,对多个位点进行敲除,编辑效率接近100%。Mans 等[57]在酵母菌中构建简单快速的敲除平台,使用含有两个sgRNA 的质粒,一步转化后可以同时引入6 个基因,促进了CRISPR/Cas9 系统在酵母中的快速、标准、高效的应用。2016 年,Shi 等[62]构建了一个新的平台Di-CRISPR(delta-integration CRISPR-Cas),用于高效、无标记、单步、多拷贝的酿酒酵母生化途径的整合,可整合长度高达24 kb 共计18 个拷贝的基因片段。2018 年,Bao 等[63]开发了一种CRISPR/Cas9 同源定向修复基因组工程(CHAnGE)的方法,可快速编辑基因组单核苷酸,超过98%的目标序列被成功编辑,编辑效率平均为82%,并利用该方法构建了一个突变酵母文库。2019 年,刘子鹤团队[64]构建了一种gRNA-tRNA 阵列CRISPR/Cas9(GTR-CRISPR)系统用于酿酒酵母的多基因编辑,该系统能以80%的敲除效率同时敲除8 个基因,极大地提高了酿酒酵母基因组的编辑效率。
这些平台的构建,使酵母合成生物学得到快速发展,并应用于许多其他种类的工程酵母菌中。2014 年,Ryan 等[61]利用CRISPRm 系统在二倍体酵母中改良纤维二糖利用途径,使得纤维二糖发酵速率提高10 倍以上。2015 年,Jakočiūnas 等[65]尝试了很多基因敲除的组合,在没有过表达甲羟戊酸(MVA)途径相关基因的情况下,得到了比野生型菌株MVA 产量高41 倍的工程菌株。2016 年,Shi 等[62]利用Di-CRISPR 平台构建了一个利用木糖生产(R,R)-(−)-2,3-丁二醇的酵母菌株,产量高达12.51 g/L。2017 年,Lian 等[66]提出了一种三功能CRISPR 系统组合代谢工程策略,即结合了酵母的转录激活、转录干扰和基因敲除三种功能,命名为CRISPRAID,这种策略能够以模块化、高通量的方式调控代谢网络,使β-胡萝卜素的产量提升了3 倍。同年,Apel 等[67]构建了一个基于CRISPR/Cas9 的无克隆工具包,该工具包包括了23 个Cas9-sgRNA 质粒、37 个表达强度和表达时间不同的启动子以及10 个蛋白质定位、降解和助溶标签,解决了酵母基因操作中选择染色体整合位点、选择启动子、蛋白质定位和溶解性等问题,利用该工具优化了紫杉醇合成酶的表达,使紫杉二烯的产量提高了25 倍。2018 年,Xue等[68]通过使用CRISPR/Cas9 技术完全敲除乙醇脱氢酶基因(ADH2),使生物乙醇的产量提高了74.7%。2019 年,刘子鹤团队通过GTR-CRISPR 平台简化了酵母脂质代谢网络,将游离脂肪酸产量提高了30 倍[64]。另外,通过CRISPR/Cas9 技术对生物乙醇[68-70]、3-羟基丙酸[71]、萜类[72]和青蒿素类[73]的生物合成过程进行编辑和增强,这些物质的产量均得到了很大的提升。这些研究说明了在酵母中CRISPR/Cas9 技术的发展和应用已经非常成熟。基于此,大量高效、创新、实用的CRISPR/Cas9技术不断被开发出来,服务于酵母合成生物学的研究与发展。
微生物合成生物学的另一个重要目标就是人工设计并合成底盘生物,元英进课题组运用CRISPR/Cas9 技术构建了人工合成酿酒酵母5 号染色体[74],拓宽了该技术在酵母中应用的外延。另外,覃重军课题组利用该技术将酿酒酵母天然的16 条染色体整合为一条具有完整功能的染色体SY14,并进一步利用该技术将其构建为环状染色体[75-76]。这些研究为CRISPR/Cas9 在微生物合成生物学中的应用和发展提供了新的思路和方向,也证明了该技术在酵母中的应用效果优于在细菌中的应用,可作为一种强有力的基因编辑工具在更多的研究和实践中大规模使用。
2.3 CRISPR-Cas9 技术在其他微生物基因编辑中的应用进展
放线菌(actinomycetes)是一类生产β-内酰胺类、四环素类、利福霉素类、氨基糖苷类、大环内酯类和糖肽类等抗生素的菌种[77]。2015 年,Cobb等[78]首次将CRISPR系统在链霉菌属(Streptomycesspecies)中应用,实现了对S.lividans的双基因同时编辑和31 kb 片段的敲除,同时构建了含sgRNA 和Cas9 相关基因的pCRISPomyces-2 温敏型质粒,为CRISPR/Cas9 技术在链霉菌中的应用奠定了基础。为了更好地服务于工业生产,在工业放线菌中CRISPR/Cas9 技术也被开发出来。2016 年,Wolf 等[79]利用该技术在游动放线菌SE50/110(Actinoplanessp.SE50/110)中敲除酪氨酸酶基因(melC2),消除副产物异黑素(different melanin)的生成,有助于提高目标产品阿卡波糖的纯度。2017 年,Jia 等[80]对S.rimosus中葡萄糖-6-磷酸脱氢酶同工酶基因(zwf2)和6-磷酸葡萄糖酸内酯酶基因(devB)进行突变和敲除,使土霉素产量增加了36.8%。2018年,Liu等[81]在红色糖多孢菌(Saccharopolyspora erythraea)中敲除SACE_1765,并在SACE_0712 处引入pErmE 启动子,使红霉素产量与野生型相比提高了80.3%。另外,还可通过敲除某些基因鉴定未知的代谢途径。例如,Low 等[82]在链霉菌SD85 中利用该技术敲除模块化聚酮合酶基因(sceN),证实了该基因负责合成抗真菌多烯大环内酰胺。
丝状真菌(filamentous fungi)是一类具有紧密基因组的真核微生物,参与一些初级代谢产物和次级代谢产物的生产,包括有机酸、酶、多糖和抗生素等,在工业和农业等领域都发挥了重要作用[83-84]。2015 年,Liu 等[85]将体外转录的gRNA和外源供体DNA,引入组成型表达Cas9 蛋白的里氏木霉(Trichoderma reesei)中,实现了使用CRISPR/Cas9技术的基因编辑,其中单基因编辑效率近100%,双基因同时编辑的效率为45%,三个基因同时编辑的效率4.2%。2016 年,Pohl等[86]在产黄青霉(Penicillium chrysogenum)中实现了负责绿色素合成的两个基因amds和pks17的突变。2017 年,Qin 等[87]首次在大型真菌灵芝(Ganoderma)中应用CRISPR/Cas9技术,有效提高了灵芝中抗癌物质灵芝酸的含量。除此之外,CRISPR/Cas9技术在其他真菌中的应用也非常广泛,如米曲霉(Aspergillus oryzae)[88-89]、粗糙脉孢菌(Neurospora crassa)[90]、稻瘟菌(Pyricularia oryzae)[91]、白色念珠菌(Candida albicans)[92]、棘孢曲霉(Aspergillus aculeatus)[28]。
噬菌体(bacteriophages)是地球上数量最多的生物,广泛存在于地球上所有的水域中[93]。噬菌体通常有一个二十面体的头部,在头部的蛋白质壳里包含了一个显性的dsDNA 基因组。除了头部以外,大部分噬菌体都有尾巴,其作用是将基因组传递到宿主细菌中,以达到增殖的目的[94]。为了更好地利用噬菌体,需要对噬菌体进行基因编辑以符合特定的生物学需求。由于CRISPR 系统源于微生物对外来噬菌体抵抗,因此将CRISPR 技术用于噬菌体基因组编辑目前存在一定困难。2017 年,Tao 等[93]首次利用CRISPR/Cas9技术对T4 噬菌体进行了基因编辑,他们将CRIPSR/Cas9 质粒和供体DNA 质粒共同导入宿主大肠杆菌,成功修饰了T4 噬菌体的基因组,为噬菌体的基因编辑提供了宝贵的经验。2019 年,Hoshiga 等[95]对T2 噬菌体的尾巴纤维进行了修饰,使T2 噬菌体对大肠杆菌O157:H7 的吸附能力与PP01 相当,对于治疗由该种类大肠杆菌引起的腹泻具有很大的意义。由上述研究可知,CRISPR/Cas9 技术在微生物合成生物学中具有广泛而深入的应用,使底盘生物的改造更加快速、准确且高效,为微生物合成生物学的发展奠定了理论和应用基础。因此,人们对于该技术有了更多的要求和研究,在此基础上延伸出了更多的基因编辑技术。
3 CRISPR/Cas9 衍生技术在微生物合成生物学领域的应用
3.1 CRISPR/Cas12a(Cpf1)技术
CRISPR/Cas12a 系统属于V 型系统,Cas12a为单亚基蛋白[96],与Cas9 蛋白相比,该蛋白具有以下特点:①识别切割DNA 不需要tracrRNA;②PAM序列富含胸腺嘧啶,通常为TTTN;③PAM序列位于被识别的DNA 的5′端;④识别点和切割点距离PAM 较远;⑤切割后产生黏性末端[35];⑥蛋白分子量更小;⑦需要的crRNA 序列更短。该系统的具体作用机制如图2(a)所示。目前,CRISPR/Cas12a 技术在常见的工业菌株如酵母[97-99]和链霉菌[100]中均有研究,并构建了相应的编辑平台(表1)。如Liu 等[101]基于Cas12a 设计了一种具有基因编辑和转录抑制双重功能的CRISPR 系统(RE-CRISPR),使谷氨酸棒状杆菌(Corynebacterium glutamicum)的半胱氨酸和丝氨酸产量分别提高了3.7倍和2.5倍。
蓝细菌(Cyanobacteria)是一种可以进行光合作用的原核微生物,蓝细菌的光合固碳和生物合成化学品是合成生物学领域研究的热点[102]。目前主要利用同源重组方法编辑蓝细菌的基因,由于其基因拷贝数较高,导致基因编辑效率极低。有相关研究表明,Cas9 蛋白对蓝细菌类微生物有一定的毒性,因此使用CRISPR/Cas9 系统对其进行基因编辑更加困难[103]。2016 年,Ungerer等[104]率先实现了利用CRISPR/Cas12a 对蓝细菌的基因编辑,实现了在三种不同类型的蓝细菌中进行基因敲除、插入和突变的基因编辑操作。2019 年,Niu等[105]在AnabaenaPCC 7120 中利用不同抗性标记多个基因组,同时使用蔗糖敏感性质粒,使其在完成编辑后主动丢失,实现了染色体大片段DNA缺失,单基因修饰达成功率100%。由此可知,CRISPR/Cas12a 系统可以实现蓝细菌的编辑基因,这些研究为CRISPR 技术在蓝细菌中大规模应用奠定了理论和应用基础。上述研究都说明CRISPR/Cas12a 编辑细胞的效率较高、特异性较高、脱靶率较低且可以编辑部分Cas9 敏感的菌种,我们可以推测其可以替代以大肠杆菌为代表的细菌中利用的CRISPR/Cas9 系统,提高细菌中基因编辑的效率和特异性。
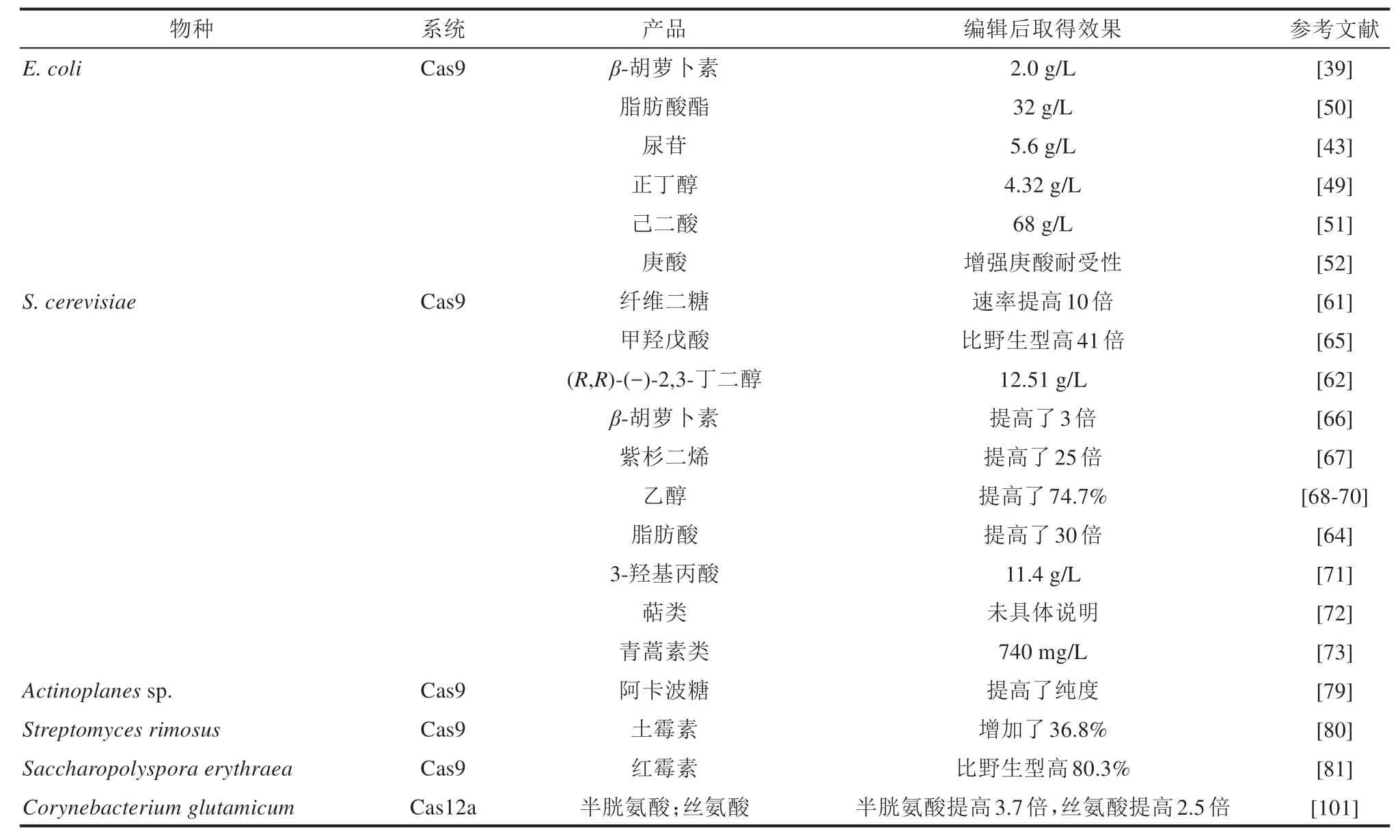
表1 CRISPR技术在微生物合成生物学中生产目标产品的研究Tab.1 Applications of CRISPR-based technologies for the construction of microbial cell factories
3.2 CRISPR/Cas13 和Cas14
除了Cas9 和Cpf1 蛋白以外,Cas13 系列的蛋白也逐渐被重视(表2)。2016 年,张锋团队在细菌中发现了一种能对抗RNA 病毒的核酸酶C2c2(Cas13a),该酶能与RNA 靶向结合并裂解RNA,实现细菌自我防御[图2(b)]。由此发展出RNA 水平上的基因编辑,可实现在DNA 不损伤的条件下对RNA 进行干扰[106]。同时,Cas13 系列中的C2c6(Cas13b)和Cas13d 也具有相同的特点[107-108]。Cas14 来源于一种古细菌的CRISPR/Cas系统,与Cas9 蛋白相比,其体积更小,且可在不要PAM 序列的情况下靶向单链DNA 并对其切割,具有广泛应用的潜力[109]。目前为止,Cas13 和Cas14 在微生物合成生物学领域研究甚少,技术并不完善和成熟。但是,在RNA 水平上的编辑是可逆的、变化的,因此,可在此基础上开发动态调控元件,调节Cas13 或Cas14 蛋白的表达量和表达时间控制通路的调节程度和调节时间,是一种非常有前景的应用方式。基于上述特点,这两项技术未来在微生物合成生物学领域将有极大的应用和发展空间。
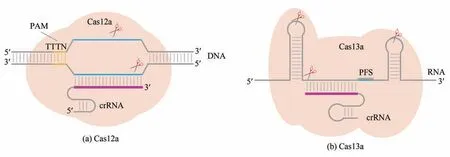
图2 Cas12a和Cas13a系统作用机制[Cas12a中橙色序列为TTTN的PAM序列,Cas13a中蓝色序列为PFS(Protospacer flanking site)序列]Fig.2 Working mechanism of Cas12a and Cas13a systems[Orange in Cas12a represents the PAM sequence of TTTN,and blue in Cas13a represents PFS(protospacer flanking site)sequence]
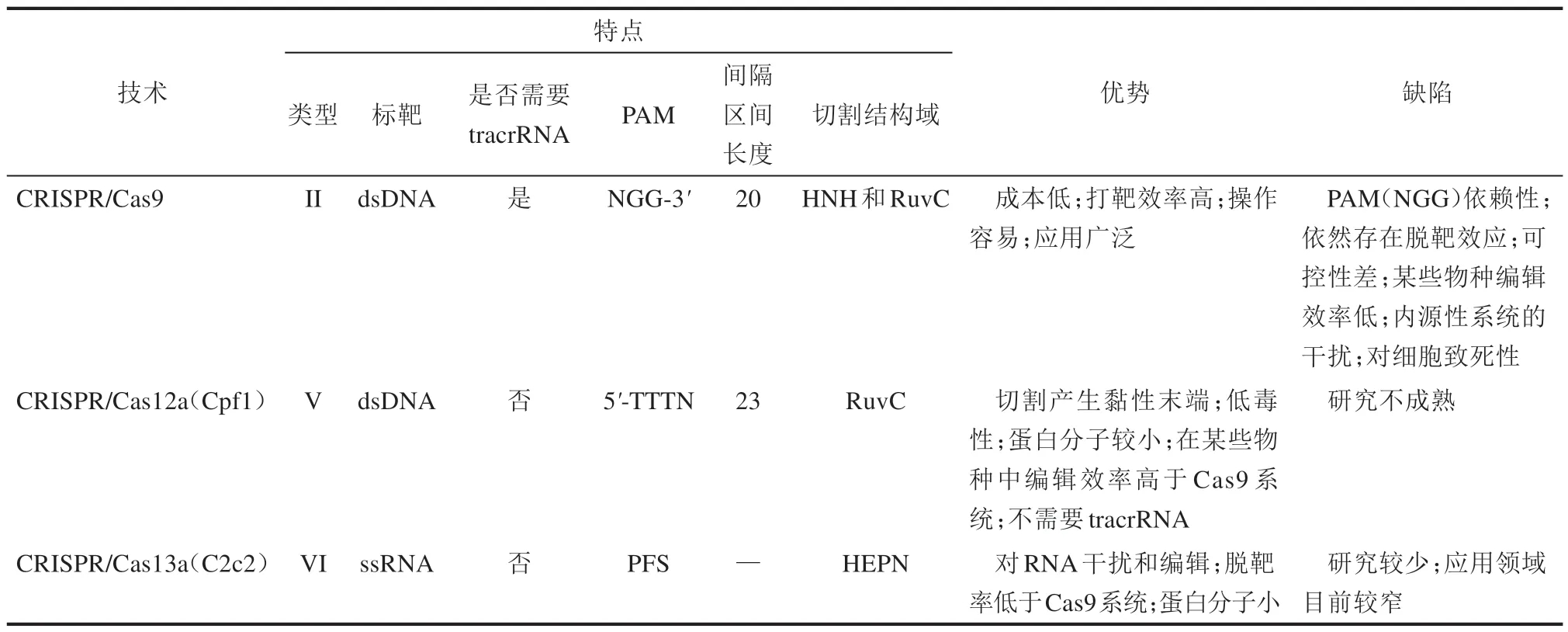
表2 CRISPR/Cas9、Cas12a、Cas13a对比总结Tab.2 The comparison and summary of CRISPR/Cas9,Cas12a and Cas13a
3.3 多亚基Cas蛋白的研究
目前,以单一亚基的Cas蛋白为核心的研究已广泛应用于基因组编辑、转录调控等不同的领域,但大多数CRISPR/Cas 系统中的Cas 蛋白是多亚基的[110]。因此,这种广泛的多亚基Cas蛋白系统在应用方面也有无穷的潜力。根据分类,多亚基的CRISPR/Cas系统包括Ⅰ型、Ⅲ型和Ⅳ型[33]。这些系统的Cas蛋白在发挥作用时,并非像Cas9等单一蛋白一样,由一个亚基的不同结构域发挥不同的作用,而是由多个不同的Cas 亚基共同发挥作用[7]。2019年,Dolan等[111]在人体胚胎干细胞和HPA1细胞中引入sgRNA,实现了远程基因敲除。该研究通过T.fusca级联和Cas3 的RNP 传递获得了13%~60%的编辑效率,表明Ⅰ型CRISPR/Cas系统在真核生物的基因组编辑和敲除方面具有潜在的用途。同年,Hidalgo-Cantabrana 等[112]利用内源性的Ⅰ型CRISPR/Cas 系统,对卷曲乳杆菌(Lactobacillus crispatus)进行了碱基缺失和插入的突变,这为重新利用内源性CRISPR系统灵活靶向和编辑基因提供了一个框架。而Ⅲ型和Ⅳ型的CRISPR/Cas系统的作用机理方面的研究已经有了突破性的进展[113-114],但应用并不广泛,在微生物合成生物学领域应用也很少。因此,由于多亚基Cas蛋白系统的广泛性,其在合成生物学领域的应用还有待发掘。
4 总结和展望
CRISPR 技术的出现极大地促进了合成生物学的发展,由于其方便、快捷、高效、成本低、难度小等特点,为编辑动植物和微生物的基因组提供了强有力的工具。但是,CRISPR 技术目前还存在一定的局限性。
(1)PAM 序列的依赖性。传统的CRISPR/Cas9 技术依赖于NGG 序列,因此无法实现对任意位点的编辑。虽然Cas12a 提供了新的PAM 依赖序列(T)TTN,在一定程度上缓解了NGG 依赖性的问题[35],但位点依赖性并没有被彻底解决。因此,提高CRISPR 技术的普适性依赖于研究出一种不需要PAM 序列的方法,或者通过理性设计研究识别基因高保守区碱基序列如TATA 或NTG 的方法。目前的研究已经发现了Cas14蛋白在靶向目标DNA时不需要PAM 序列[109],因此Cas14具有解决CRISPR技术对依赖PAM序列的潜力。
(2)编辑的脱靶率。研究者们为了解决脱靶问题分别从sgRNA和Cas蛋白入手进行研究。张锋团队建立了gRNA-Cas9n系统,Cas9n蛋白切割时只产生一个切口,通过两套gRNA-Cas9n 系统同时对目标基因切割,可提高sgRNA的特异性[115],降低脱靶率。相关研究表明富含鸟嘌呤并缺乏腺嘌呤的sgRNA 更稳定,由此开展了对sgRNA 活性的研究[116]。通过大规模数据的收集并研究sgRNA 的活性,Guo等建立了一个全面的sgRNA活性图谱,可在模式生物任何位点选择出高活性的sgRNA 序列,有助于提高sgRNA 的特异性并降低脱靶率[117]。同样,对Cas蛋白的改进研究较多,如使用FnCpf1[118]等,这些研究有望解决CRISPR技术脱靶率的问题。
(3)该系统的安全性控制问题。2017 年,Shin 等发现了抗CRISPR 蛋白Acr II A4 对CRISPR/Cas9 系统的抑制作用[119],为精准控制CRISPR 系统奠定了基础,也为控制该系统带来了希望。
(4)应用广泛性。虽然CRISPR 技术已经广泛应用于多种微生物系统,但在某些微生物中依然难以应用。Cas9 蛋白对蓝细菌等具有毒性,因此CRISPR 技术很难在蓝细菌中应用,虽然Cas12a可以应用于蓝细菌中,但是Cas9 蛋白对细胞产生毒性而Cas12a 蛋白的毒性较低的原因依然没有较好的解释。而这些问题和困难的不断解决会促使CRISPR 基因编辑技术在微生物合成生物学领域具有更加广泛的应用,从而可以快速、高效、精准地创造出更多适合生产高附加值产品的底盘生物。无论是微生物合成生物学还是CRISPR 技术都是近年来发展起来的新兴学科和技术,两者都有很多问题等待研究和解决,随着两者相辅相成地快速发展和深入研究,在未来一定会得到更多的研究成果和更好的技术应用。
猜你喜欢
军事文摘(2022年16期)2022-08-24
麦类作物学报(2022年3期)2022-05-19
临床与实验病理学杂志(2022年3期)2022-04-06
中国饲料(2021年17期)2021-11-02
今日农业(2021年11期)2021-08-13
现代畜牧科技(2021年4期)2021-07-21
世界最新医学信息文摘(2020年68期)2020-12-25
中国生殖健康(2020年4期)2020-12-09
中西医结合肝病杂志(2020年2期)2020-10-27
烟台大学学报(自然科学与工程版)(2020年1期)2020-02-08
