
蛋白质计算设计:方法和应用展望
2021-03-18 07:52操帆陈耀晞缪阳洋张璐刘海燕
合成生物学 2021年1期
操帆,陈耀晞,缪阳洋,张璐,刘海燕
(中国科学技术大学生命科学学院,安徽 合肥 230026)
蛋白质是执行生物功能的主要生物大分子,也是用于构筑合成生物系统的主要元件。大多数蛋白质的功能取决于它们的特定三维空间结构和特异性分子间相互作用。氨基酸序列决定了蛋白质三维结构和相互作用,从而决定蛋白质功能。天然蛋白质的氨基酸序列经过了进化的长期选择,适应了相应有机体的功能需求。在合成生物学中,当天然蛋白被转用于其他目的时,其性质和功能很可能达不到要求,有时甚至找不到可用的天然蛋白。因此,对天然蛋白的性质和功能进行定向改造,乃至创造有新功能的人工蛋白,对合成生物学具有重要意义[1-5]。传统蛋白质工程技术如定向进化[6]对天然蛋白序列进行小的扰动,本质是一种试错方法,在不采用高通量筛选手段时效率很低,且难以创造出具有新结构和新功能的蛋白。因此,经验的或计算的蛋白质理性设计成为了改造乃至创造新蛋白质的手段。其中,依赖经验知识以及进化信息等[7]的理性设计在改造蛋白质方面确实有一些成功案例,但是难以解决复杂的蛋白质的工程问题。蛋白质计算设计,即从结构功能的需求出发,通过计算手段确定氨基酸序列[8-11],既可以用于蛋白质从头设计,也更多地被应用于既有蛋白质的改造设计,是亟待推动的发展方向。目前,以蛋白质从头设计为目标开发的一些计算方法已被越来越广泛应用于蛋白质工程改造中。有报道表明,在蛋白质相互作用界面改造中,通过计算设计技术的恰当应用,可以把实验试错范围缩小3~4 个数量级[12]。目前,计算设计方法还有巨大的发展空间,且相关研究队伍也日益扩大。计算方法不仅会在对天然蛋白的理性改造中得到广泛实际应用,按需定制的人工设计蛋白的实际应用也有可能在未来5~10 年内普遍实现。
本文主要目的是介绍目前已采用和正在发展中的蛋白质计算设计方法的基本原理,面临的主要问题和解决思路、方法,以及尚待解决的一些问题和可能的研究方案。尽管这些方法的最终发展目标是蛋白质按需从头设计,它们也易于被调整用于蛋白质理性改造。在下文中,本文作者首先简要回顾蛋白质计算设计的发展历史,认识该领域现状和整体发展方向;随后主要围绕蛋白质从头计算设计,介绍其基本方法和原理;并汇集总结一些具体应用成果,讨论蛋白质从头计算设计应用的主要思路;最后对方法发展和应用的趋势进行简要展望。
1 蛋白质计算设计的历史
本节主要从设计策略的角度,对采用不同类型策略的方法分别概述。尽管多种方法被首次报道的时间较早(如20 世纪80 年代出现的基于规则的启发式设计方法、20 世纪90 年代出现的通过自动优化能量函数进行序列设计的方法),但直到今天它们仍在持续的应用、验证和完善中。对相关时间顺序感兴趣的读者可参考其他综述[13]。
1.1 基于规则的启发式设计
最早被提出的蛋白质设计方案受到了特殊的、高度规则的多肽结构的序列变化规律的启发[14-16]。多肽主链高度规则的局部结构模式包括α-螺旋、β-片层等二级结构单元。多个二级结构单元之间能够以特殊方式相互堆积扩展成更大的三维结构单元,如超二级结构motif、多螺旋束等。与之对应的氨基酸序列上,不同性质氨基酸呈现特殊的排列模式,如图1 中反平行螺旋上A、D 位置由疏水氨基酸占据,其余位置则多被亲水氨基酸占据;β-肽段上亲、疏水氨基酸周期性地相间排列,以使疏水侧链埋于蛋白质内部,亲水侧链暴露在溶剂中。基于这种排列模式设计氨基酸序列的启发式方法被成功应用于设计各类螺旋束结构[17-18]、超二级结构motif[19]等,其中发展较为系统的是多螺旋束设计。为了更系统地刻画多螺旋束中不同螺旋间堆积结构可能的变化,Grigoryan 和De Grado 等[20]建立了精细的经验数学公式来定义螺旋间距、扭转角、相对平移等几何参数间的相互依赖关系,用于设计不同数目和排列的理想螺旋束结构。这类设计方法也存在着明显的局限,首先它受限于特殊、有限的主链结构类型;此外,仅仅通过区分残基亲、疏水性等经验来选择残基类型得到的设计结果具有很大的不确定性,由于没有控制残基之间特异性的空间堆积和氢键相互作用等,最终获得能特异性折叠序列的成功率并不高。
1.2 通过自动优化能量函数进行的序列设计
20世纪90年代后期,随着分子力学能量函数、氨基酸侧链构象库、优化算法等的发展,Dahiyat等[21]首先实现了用自动优化的方法来设计氨基酸序列。在此类算法中,主链骨架是被事先给定的(如来源于天然蛋白质结构),且可被假设为固定不变。设计中需要通过计算来确定的未知量包括每个主链位置上的氨基酸残基类型以及其侧链构象。这些未知量的所有容许取值(即氨基酸侧链类型及其构象状态的可能组合)构成了氨基酸序列和侧链构象空间。定义在该空间上的能量函数则被用于评估特定序列和构象组合的好坏。定义了主链结构和能量函数后,设计者通过特殊的算法在序列和侧链构象的未知量空间中自动搜索,找出能量尽可能低的解,得到设计结果。图2 简要演示了这一设计过程,对于左侧输入的目标主链结构,通过搜索序列和侧链构象空间,找到具有最低能量的序列,认为它们就是最可能形成目标结构的序列。值得一提的是,实现这类设计算法的关键技巧之一,是将本来连续变化的侧链构象离散表示为可数的有限种可能状态(称为rotamer)。设计算法的另一关键是能量函数。从原理上,如果能找到普适的能量函数,基于能量函数自动优化的设计方法就能被广泛应用于不同结构类型蛋白的设计。因此,从被提出至今,通过优化能量函数进行自动设计逐渐成为蛋白计算设计的主流策略,而相应的能量函数[22-24]和优化算法[25-26]等得到持续的发展。到目前为止,至少两套能量函数(Rosetta 能量函数[25]以及本文作者课题组建立的ABACUS 统计能量函数[27-28])都已被实验反复验证能以很高的成功率进行氨基酸序列从头设计。以天然主链结构为设计目标,用ABACUS 进行氨基酸序列全自动设计得到的人工蛋白往往具有远超天然蛋白的高热稳定性[27]。
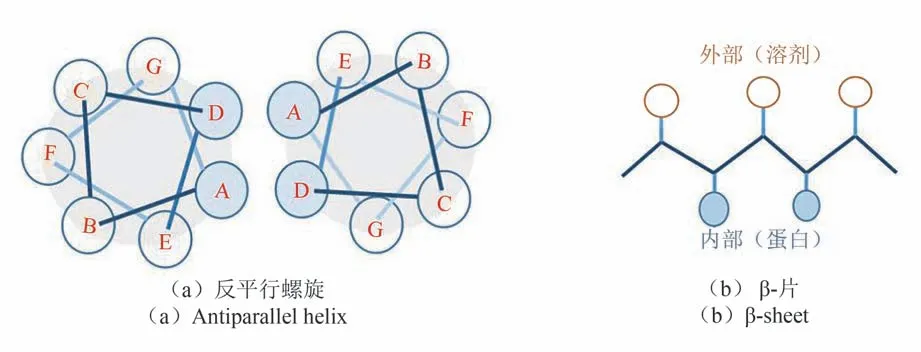
图1 形成规则空间结构的多肽链的氨基酸序列变化规律示例Fig.1 Examples of changes in the amino acids sequence of a polypeptide chain forming a regular spatial structure
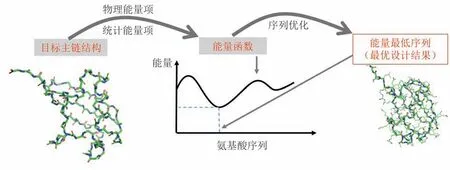
图2 给定主链优化氨基酸序列和侧链构象Fig.2 Optimization of amino acids sequences and side-chain conformations for a given backbone
为了把计算量控制在可行范围内,在优化氨基酸侧链类型和构象时,主链结构一般被假设为固定不变的。如果主链结构也被作为未知量与序列、侧链同时被优化,尽管物理层面上更合理,但计算层面上,变量空间维度会过高,使得计算无法完成。另外,对主链结构难以进行合理的离散采样,对其进行优化比固定主链优化侧链类型和rotamer 的组合要困难得多。为了在一定程度上考虑主链柔性,研究者提出了不同的方案,其基本思路都是对多种互有差别的主链结构进行序列设计。应用最多的方案是在序列空间和主链结构空间的优化交替迭代进行,这是多数Rosetta Design 应用中采取的方法[29]。另一种方案是对多个主链结构的集合(主链系综)同时优化氨基酸序列[30]。研究者提出了不同方法产生主链结构系综,以尽可能合理再现在天然同源蛋白中观察到的序列差异引起的主链结构的可能变化,如所谓的“backrub”运动[31]等。值得指出的是,这些方案不是对主链构象空间进行大范围采样。它们仍然需要从一个与最终结构非常接近的初始主链结构模型出发。最终结构只是初始主链附近的小幅度变化(主链原子均方根位移最大在1~1.5 Å 左右)。是否以这种方式处理主链柔性似乎对主要基于统计能量函数的ABACUS 方法的设计成功率影响较小[32]。
1.3 多肽主链结构的从头设计
真正的从头蛋白质设计不应仅限于用天然主链结构作为设计目标。满足最基本化学要求(共价构型正确、原子间无空间冲突)的可能主链构象是非常多样的。其中占比非常少的构象才具有所谓的“可设计性”,即存在氨基酸序列,能自发稳定地折叠成这种构象。从头设计的主链结构必须具有高“可设计性”。如何保证这一点,到目前为止,还没有经实验充分验证的普适方案。目前成功例子最多的,是通过引入结构预测中使用的算法来形成问题特异的启发式方案。这类方案的基本步骤为:定义要设计的目标主链结构的基本框架(二级结构单元的组成、大致相对位置等),产生对主链结构的约束条件;再把天然蛋白质中的主链结构片段和二级结构元素拼接成满足约束条件的初始结构;进而用结构预测中使用的能量函数、构象采样方法进行结构优化,进入主链结构/序列设计的优化循环。为提高人工构建主链结构的可设计性,Koga等[33]分析了二级结构模式和三级结构模体之间的关联性,统计了不同空间连接方式的二级结构单元间环区长度和构象分布,提出了如何设计环区长度和构象的经验规则。目前用这种方法人工设计的主链结构在二级结构及其连接区等局部结构特征上大多具有理想的结构模式,缺乏天然蛋白展示出的主链结构的丰富多样性[34]。此外,主链结构优化时使用全原子能量函数,依赖于侧链类型和构象,故而通过主链优化-序列优化迭代的方式进行设计。除了利用天然主链结构和序列片段拼接设计人工蛋白外,Frappier 和Mackenzie 等还提出通过分析天然蛋白三维结构数据库,定义空间相邻的多个短片段构成的三维结构单元(称为TERM),用TERM 的组合来进行蛋白质设计[35-36]。另一可能的解决方案是构建不依赖于侧链类型的主链能量模型,直接通过主链能量优化进行主链设计[34,37-38]。
2 蛋白质从头设计的计算方法
本节从以下四个方面来介绍蛋白质从头设计的计算方法:氨基酸序列设计;主链结构设计;蛋白质分子间相互作用界面设计;以及负设计。前两个方面前文已提到;通过针对性调整序列和结构设计方法,可为蛋白质设计新的分子间相互作用界面,从而实现新的功能;负设计是一种概念性的设计思路,将在本节最后予以补充。
2.1 氨基酸序列设计方法
对于在给定目标主链结构下进行序列设计的问题,我们通过定义能量作为序列的函数,把序列设计问题转化为在序列空间中找到能量最低的序列的最优化问题(图2)。这里,能量函数是优化问题的目标函数,它定量评估不同序列与给定目标主链结构匹配的程度:能量越低的序列越有可能稳定地形成与目标一致的主链结构。
2.1.1 序列设计的能量函数
序列设计的能量函数具有经验的数学形式,其中既有基于物理原理的能量项,也有通过对蛋白质数据库进行统计分析得到的能量项。以现在应用成功的例子最多、使用最广泛的蛋白质设计软件Rosetta[25]为例,其能量函数是刻画不同物理相互作用的能量项和部分统计能量项的线性组合,Etotal=∑iwiEi(θi,aai)。该函数中的不同能量项是基于对各种分子相互作用、对蛋白质折叠的重要性的分析和既有认识经验性地提出来的。其中物理能量项主要包括共价结构、范德华相互作用、静电相互作用和氢键、溶剂化自由能等。此外,总能量中还包括依赖于主链二面角、rotamer 类型等的统计能量项。
(1)物理能量项
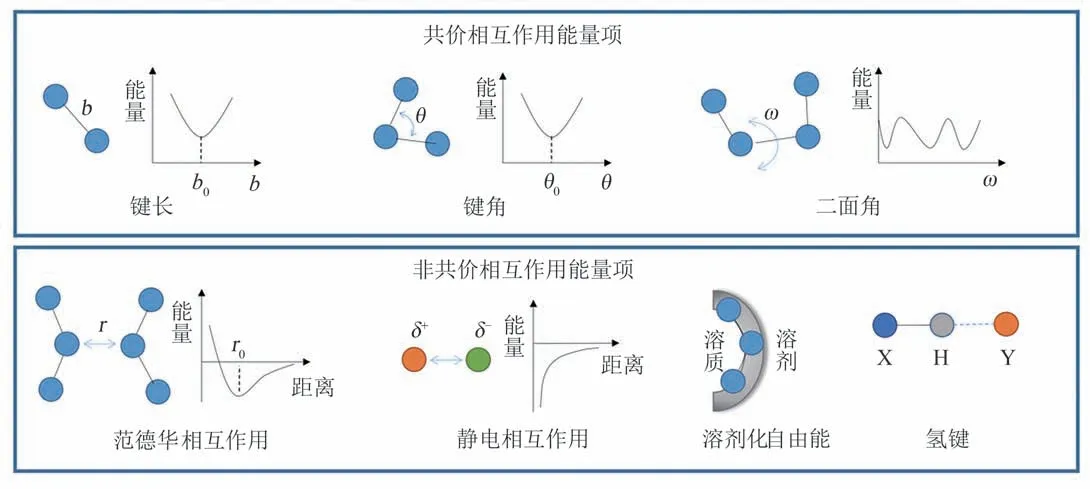
图3 物理能量项Fig.3 Physical energy terms
用于刻画蛋白质等生物大分子体系的物理能量项可分为共价相互作用能量项(键长、键角、二面角等)和非共价相互作用能量项(范德华相互作用、静电相互作用,溶剂化自由能、氢键等)两类(图3)。在序列设计中,键长、键角以及决定立体构型的非正常二面角等几何性质通常保持固定不变,共价相互作用能量项可视为常数。可变的物理能量项中,范德华相互作用能量项是随原子间距离而变化的短程排斥和长程色散吸引的加和。Rosetta 使用了吸引和排斥可拆分加权的Lennard-Jones 势来计算范德华相互作用能量。静电项刻画带电的极性官能团之间的库仑相互作用,Rosetta使用最初来自CHARMM分子力场的原子电荷分布来计算静电能,并通过组优化进行了调整。氢键是亲核重原子将电子密度提供给极性氢时形成的部分共价相互作用。Rosetta 使用了静电模型和特殊的氢键模型来计算氢键的能量,并且该能量被细分为不同的类型分别计算:长距离主链氢键、短距离主链氢键、主链和侧链原子之间的氢键、侧链之间的氢键。溶剂效应在决定蛋白质构象时发挥了至关重要的作用。分子能量函数中常用的溶剂模型分为显式溶剂模型和隐式溶剂模型[39]。显式溶剂模型需要对每个溶剂分子的原子空间位置进行采样并据此计算溶质-溶剂原子间的相互作用。由于计算量较大,显式溶剂模型对序列设计是不合适的。隐式溶剂模型则通过定义只依赖于溶质结构坐标的有效溶剂化自由能来处理溶剂效应。Rosetta 中使用的Lazaridis-Karplus(LK)隐式高斯排除模型[40],溶剂化自由能包括各向同性的溶剂化能量以及各向异性的溶剂化自由能两部分,分别刻画非极性和极性溶剂化效应。
(2)统计能量项
统计能量项是对数据进行统计分析得到的概率分布进行转化后得到的(图4),通过对数据库中不同的构型变量分布进行统计分析,将其出现的概率转换为能量,对依赖于多个几何变量的高维统计能量项(例如依赖于构象和环境的主链位点之间的相互作用),需要发展特殊的技术,才能恰当地估计多维概率密度,从而得到合理的统计能量函数。可以从两个不同角度来理解序列设计的统计能量项。一是从统计热力学角度,在平衡态,物理系统处于不同微观状态的概率服从玻尔兹曼分布,。其中r代表微观状态的坐标,E(r)代表微观状态的能量,kBT是玻尔兹曼常数乘以环境的热力学温度。因此,我们可以根据数据集合中的概率分布反推出相互作用E(r)=-kBTlnp(r)+常数。另一个角度则可从纯统计学角度出发,假设给定主链结构后氨基酸序列分布可记为条件概率P(sequence|backone),序列设计要解决的问题是寻找让该条件概率最大的序列。如果我们定义统计能量E=-ln(p),则概率最大化等价于能量最小化。

图4 不同类型的统计能量项Fig.4 Statistical energy terms of various types
需要注意到,无论是微观状态的概率P()r,还是条件概率P(sequence|backone),都是非常高维的函数,其变量的各个分量之间高度互相依赖。是无法从数据集中直接估计出这样的高维概率分布的。通过应用玻尔兹曼分布的反转,我们把对概率分布的估计转化成对自由度之间相互作用的估计,从而可以在统计时对自由度之间的耦合分类处理,只保留较低阶的耦合对总能量的贡献,以把问题复杂程度控制在可处理范围内。换一种说法,全序列的总能量,被分解为由序列上每个位点的残基类型分别决定的单残基能量,以及刻画残基间两两相互作用的残基间相互作用项。依赖于两个以上位点的残基的更高阶能量项,则会被忽略。
这样的残基类型依赖的统计能量,可以和物理能量项加权组合起来,用于弥补物理能量项的不足。Rosetta 总能量中就使用了多个这样的单残基统计能量项,包括反映Ramachandran 主链二面角对残基类型影响的能量项、侧链构象依赖的能量项等。此外,Rosetta 还使用几何参数的统计概率分布来计算半胱氨酸形成的二硫键的能量。值得注意的是,在用这种方法考虑统计能量项时,我们假设了不同结构特征(如主链二面角、溶剂暴露程度、二级结构类型等)对残基类型的影响是相互独立、可互相加和的。这个假设实际上是不成立的,它对统计能量函数带来的不利影响可能比较大。
本文作者课题组提出的ABACUS 方法[27-28],使用了主要基于统计能量项的能量模型来进行序列设计。其主链结构依赖的能量被分解为单残基项和残基间两两相互作用项的加和。这两类能量项都是通过直接统计在给定主链结构特征的前提下的氨基酸侧链类型或类型组合的概率分布得到的。不同于以往的统计能量项,ABACUS 把不同结构特征组合起来,作为决定氨基酸类型概率分布的联合条件,单残基能量项由氨基酸所在位置的二级结构类型、Ramachandran 主链二面角、溶剂可及性面积这些特征同时决定;而残基间相互作用项则在同时考虑两个主链位点的上述结构特征之外,还考虑位点间的相对位置(包括距离和取向),把所有结构特征作为影响残基类型组合概率的联合条件。除主链依赖的残基类型能量外,ABACUS 总能量中还包括了主链构象依赖的rotamer 能量以及原子间空间堆积能量。它们是通过对天然蛋白侧链构象分布、原子间距离分布分别进行统计得到的。
(3)确定不同能量项的权重
上述把不同类型能量项组合起来构成总能量的方案是一种经验选择。参与组合的不同能量项可能反复、冗余地包括了同一物理因素的贡献(比如除范德华相互作用外,主链构象、侧链构象等能量项也会包括范德华相互作用的贡献)。对各能量项引入待定权重能一定程度抵消这种冗余计算的不利影响。另外,把这些权重作为可调参数来拟合实验数据,我们还可能把实验数据中包含的一些其他信息笼统地引入模型中,从而改善模型。目前,用实验数据训练优化权重最有效的方法是最大化天然序列恢复比例。其基本思想是使用能量函数重新设计天然蛋白质的序列,检查各位点重新设计的氨基酸残基类型与天然残基类型是否一致。在实际应用时,我们可以基于待优化的权重的特点对这一基本思路进行调整。例如:保持氨基酸序列不变,只优化各位点的rotamer 类型,检查预测的侧链构象和天然构象的偏差;只重设计一个位点的残基类型,保持其他位点的天然残基类型不变(单位点设计),等等。
2.1.2 序列和侧链构象空间的搜索和优化算法
定义能量函数后,序列设计的下一步是确定总能量最低(或尽可能低)的氨基酸序列。由于总能量还依赖于侧链构象,该搜索优化过程同时确定侧链类型和rotamer。现在已经有多种方法来解决此问题,包括确定性优化算法(例如死端消除、平均场优化)以及随机优化算法(如模拟退火、遗传算法)[41]。
确定性优化算法可以解决全局最小的问题。但是蛋白质设计搜索空间沿多个维度(即序列空间,侧链构象空间,骨架构象空间)迅速增大、物理模型可能太复杂等,可能导致确定性优化算法无法应用。确定性优化算法例如死端消除法经常应用于较小的蛋白质或少数位点的氨基酸残基类型优化问题。但是确定性优化算法在最近也有一些大的改进,例如蛋白质设计的CLEVER 算法[42],该算法建立在Keating 实验室以前开发的簇扩展算法[43]的基础上。用于蛋白质设计的簇扩展是一种将复杂的三维原子级能量函数(是原子坐标的函数)映射到仅依赖于序列的简单线性函数的技术。因此,簇扩展将输入的物理能量模型映射为一个简单得多的模型,然后可以使用整数线性规划求解器来有效地找到新模型中的最佳序列。蛋白质设计软件OSPRREY3.0 使用基于成本函数网络(CFN)处理的最先进组合优化技术,使找到全局最小序列的计算过程加速了几个数量级[44]。
相对于确定性优化算法,随机优化算法实现更为简单。尽管随机优化只是找到能量尽可能低的序列,不保证得到全局最优解,但考虑到能量函数本身并不是百分之百准确,并且能正确折叠成目标结构的序列不唯一,随机优化找到的低能量序列和真正的全局能量最低序列实际上是同等有效的。用Monte Carlo 模拟退火进行随机优化的简单算法为:从随机选择的初始序列出发;计算当前序列能量,每步随机突变一个或多个位点的残基,计算能量变化;根据能量变化值和Metropolis 判据决定接受或拒绝突变;反复迭代该步骤,至能量不再降低。使用Metropolis 判据导致降低能量的突变均会被接受,而使能量升高的突变有一定概率会被接受。该判据中使用“温度”作为参数来度量能量变化的大小。选择高温参数时能量被容许发生大的涨落,而低温时能量降低到局部极小值附近后涨落很小。在模拟退火优化中,模拟从高温开始,以消除初始序列中大范围的不合理成分,然后逐步降低温度,以更精确地确定能量极小序列。
2.1.3 考虑主链结构的柔性
相似但不完全一样的氨基酸序列折叠形成的稳定主链结构也是相近的,但不完全一样。在序列设计中考虑主链骨架柔性,可能可以增加设计结果的多样性,找到更多能满足设计目标的结果。另外,由于能量计算依赖于结构,如果能精细处理与序列改变相对应的主链结构变化,可以更精确地计算给定氨基酸序列的能量。后者对准确设计分子间相互作用界面可能非常重要,因为对分子间特异性识别非常重要的氢键、盐桥等特异性相互作用更精细地依赖于三维结构。目前还没有各方面都比较好的处理主链结构柔性的方法,现有处理方法可分为考虑单一主链构象态的柔性扰动的方法,以及基于多主链结构设计序列的方法。
(1)考虑对单一主链构象态柔性扰动的方法
受实验观察到的蛋白质晶体结构中主链构象局部涨落模式的启发,Davis等[31]提出了一种主链原子协同变化模式,称为backrub。在该模式下,相邻三个残基的主链原子的坐标变化依赖于同一个参数。在Rosetta 全原子力场的背景下,Smith 等[45]研究了使用backrub move 来进行构象采样的方法。Frappier等[35]也同样利用这一方法来设计与特定配体结合的蛋白质。在设计过程中,他们考虑配体相对于蛋白质的可能旋转和平移,同时考虑蛋白质主链原子的协同运动,将这些对蛋白质和配体坐标的操作结合起来,称之为coupled moves。为了考虑氨基酸侧链的改变,他们根据主链构象变化,计算移动的主链片段上每个潜在突变或侧链构象的能量变化,根据Boltzmann 分布计算每个潜在突变或侧链构象的概率,用于选择侧链构象。
(2)基于多主链结构设计序列的方法
这类方法常常被称为基于结构系综的设计方法,这里“系综”是指多个主链结构的集合。按统计热力学理论,同样的序列能够形成的主链结构并不是唯一的,只是不同的主链结构具有不同的概率。系综方法用多个主链结构来代表目标结构的概率分布,同时优化序列处于多个目标结构状态的能量,因此又被称为多状态设计。由于计算量较大,可包含在系综中的主链构象数目一般不能太多。在蛋白质与小分子配体界面设计中,基于对结构柔性的考虑,Lanouette 等[46]通过构建主链结构系综进行多状态设计来预测SMYD2 蛋白的底物识别空间。除此之外,Hilpert 等[47]开发了一种新的多特异性算法,即设计能与不同配体分子结合的单个目标蛋白。在该算法中,处于复合物状态的蛋白质刚开始被冗余设计为具有不同的序列;随着设计推进,越来越多的位置被根据前期设计结果约束为相同的残基类型占据,从而使设计结果逐步收敛到单一序列;最后通过贪婪选择算法(greedy selection algorithm)进行最终单一序列优化。
2.2 主链结构设计方法
主链结构设计方法可分为两大类(图5)。一类是启发式的主链设计方法,它使用天然片段进行拼接,拼接时可用参数化的模型去约束整体结构,搭建出原子水平的主链模型,然后再用原子水平的能量函数进行主链优化。因为使用原子水平的能量函数,优化时需要考虑侧链的原子,所以是在预设侧链的基础上进行迭代设计。另一类是使用不依赖于侧链的能量函数进行主链设计方法,这类方法可用于在序列待定的条件下进行主链结构的采样和优化。
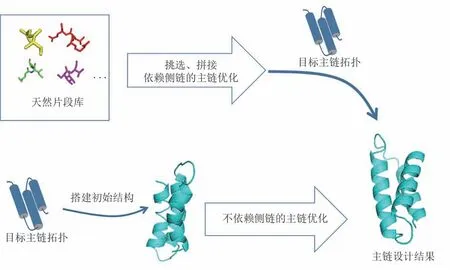
图5 两种主链设计策略Fig.5 Two backbone design strategies
2.2.1 启发式的主链设计方法
保证主链的局部结构具有高“可设计性”的一种常用方法是用天然存在的蛋白质片段来拼接组装新的主链[48],除了提供良好的二级结构之外,这些片段还可以包含在二级结构的起始和终止处高可设计性的结构模式。此外,对结构单元之间的堆积可采用参数化的模型:通过少量的参数来描述经验观察到的各类蛋白质结构单元之间的堆积特征,用于对片段拼接产生的主链结构进行约束。基于特定结构的参数化模型,可以快速生成大量蛋白质骨架。值得一提的是,这种方法对于卷曲螺旋蛋白(由围绕超螺旋中心轴的两个或多个α-螺旋组成)的设计特别适用,最新应用包括跨膜蛋白[49-50]和α-螺旋桶[51]的从头设计等。这种启发式的主链设计方法的优点在于简明,适用于设计理想的主链结构。然而也正因为使用了天然结构片段,它难以用于设计复杂的、非理想的主链结构。
Rosetta 作为一种启发式的主链设计方法使用了序列能量和主链能量相耦合的全原子能量函数,这意味着只有在假设序列已给定时才能进行主链设计与优化,因此Rosetta 实际采用预定序列的迭代策略(假定序列-优化主链-重新设计序列-优化主链)进行优化,这增加了对计算量的要求。
2.2.2 使用不依赖于侧链的能量函数进行主链设计
根据上述关于启发式的主链设计方法的分析,若能设计出一个通用的不依赖于侧链的主链能量函数,则在设计主链时将会更加自由。构建这类能量模型的途径之一是将前述统计能量函数的原理应用于天然蛋白结构数据库。早期,MacDonald等[52]发展了基于α-C 原子的能量函数来模拟主链的局部构象(即一段连续残基的主链构象)。在不依赖侧链的条件下,此能量函数的一些低能量结构仍与实验结构相似,说明能量高低能在一定程度上反映可设计性高低。该模型在描述序列上距离较远的主链堆积时使用了非常简单的函数,因此其用于优化完整主链时结果与实际主链结构的差别比较大,不适用于下一步的序列设计。我们在稍早的工作中,报道了一种称为tetraBASE 的统计能量,可以用于优化二级结构单元之间的主链堆积[11]。该能量模型假设这种空间堆积相互作用依赖于二级结构类型、残基主链的相对取向以及原子间距离。计算结果表明,在不指定二级结构单元的氨基酸序列的情况下,通过Monte Carlo 模拟退火优化不同二级结构单元之间的相对位置,可以原子水平均方误差1.5~2.5 Å(1Å=10-10m)的精度再现天然蛋白中二级结构的三维排列。这说明基于优化统计能量函数得到高可设计性的原子水平的三维主链结构模型是可能的。然而,tetraBASE 能量函数不是连续、解析可导的,它也不包含描述二级结构单元内部柔性或环区构象的能量项,用它还无法实现主链完全柔性的构象设计。最近,我们建立了一套完整描述柔性主链结构的统计能量函数,其中侧链主要作为空间位阻的保持者参与其中,因此只需使用简化的序列即可进行主链的采样和优化。我们把这个模型称为
SCUBA(side chain unspecialized backbone arrangement,待发表)。SCUBA 使用神经网络能量项来反映在高可设计性结构中多种几何结构参数间的相互依赖关系,同时保证能量对原子坐标是连续解析可导的,从而适用于随机动力学模拟等成熟的分子构象模拟采样方法。在初步验证中,我们已得到一例实例,用SCUBA 设计主链后再用ABACUS进行序列设计,得到的蛋白质实验结构符合预期(待发表)。SCUBA 提供了一种新的、在序列全部或部分待定的情况下对高可设计性主链结构进行采样和优化的方法。用SCUBA 进行结构设计可充分考虑主链柔性,从而可能推动配体结合蛋白、酶、蛋白相互作用界面设计等功能蛋白设计的发展。
2.3 蛋白质分子间相互作用界面的设计方法
一种蛋白质的功能在很大程度上是由它与其他特定蛋白质或特定小分子的特异性识别所决定的。把蛋白质从头设计的基本算法进行一些针对性的调整后,可应用于设计特异的分子间相互作用。目前已有一些设计成功的例子报道,尽管大多数从头设计的分子相互作用的亲和力相对于天然相互作用而言还不是太高。
2.3.1 设计蛋白质-蛋白质间的相互作用界面
这类界面一般位于蛋白质表面。设计的基本步骤如图6所示,首先设计与目标受体(绿色)形成复合物的配体蛋白的主链构象(红色),再设计和优化配体蛋白界面的残基类型,从而得到最终设计结果(蓝色)。设计复合物主链结构时,要考虑的首要特性是两个表面几何形状的互补性。如果要从头设计新的相互作用界面,这个性质可在表面残基类型待定的前提下,用来指导分子对接等算法,确定两个分子主链骨架之间的相对位置和取向,即复合物的主链结构。如果是对已有复合物界面进行序列重设计,则可以使用原始复合物的主链结构。总体而言,复合物主链结构设计采用启发式分子对接的方案居多,尽管目前采用这些方案能得到的界面往往达不到预期的相互作用密度[53]。
在确定复合物主链结构后,可以用自动优化的方法重新设计界面处的氨基酸序列[18]。界面序列设计的一个主要困难是界面残基间的相互作用既包括疏水相互作用,也存在大量氢键、盐桥等极性相互作用。其中疏水相互作用对亲和力的绝对贡献很大,但缺乏特异性。而极性相互作用是保证相互作用特异性的主要因素。关于蛋白质分子间界面残基分布的一个流行的模型是“O型环”,环的中心是疏水残基紧密堆积形成的核,该核被极性相互作用残基环绕。目前,对残基间极性相互作用设计的准确度还不高。如何利用界面的各类序列特征从头设计亲和力和特异性媲美天然界面的人工蛋白相互作用界面,仍然是十分大的挑战。另一种设计思路,是把天然蛋白质复合物中反复出现的界面结构模式“移植”到其他表面。比较典型的是平行或反平行堆积的螺旋产生的蛋白界面。这样的界面多肽主链结构规则,残基侧链间形成的规则氢键网络被成功“移植”的可能性更高。
2.3.2 设计小分子配体识别口袋
对酶、别构蛋白等,小分子配体结合口袋是其功能中心。特异性识别口袋的设计是功能蛋白质设计的重点。一种“由内向外”(inside-out)的基本设计思路是[9]:首先设计一个或多个由围绕目标配体的孤立残基组成的虚拟口袋结构,这些残基的位置和构象使其能够以最有利的方式与配体发生相互作用;下一步是用虚拟口袋筛选能够提供这样一个口袋结构的蛋白质骨架(RosettaMatch 算法假设给定主链骨架不变,找到能与构成虚拟口袋的残基位置达到最佳几何匹配的一组骨架位点[54]);接着,通过筛选大量主链骨架,得到最佳匹配的主链骨架以及相应的口袋残基定位组合;最后,把虚拟口袋转移到筛选出的蛋白骨架中后,可对口袋附近的残基再进行重新设计和优化。
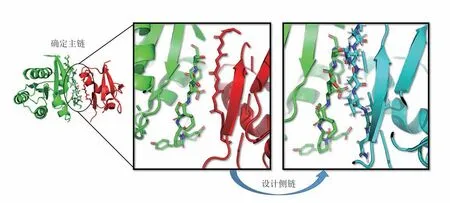
图6 蛋白质-蛋白质界面设计的基本步骤Fig.6 Basic steps of protein-protein interface design
2.3.3 设计氢键网络
无论是蛋白质-蛋白质相互作用界面还是小分子结合口袋,分子间氢键网络对在保证高亲和力的同时维持相互作用的高特异性具有重要意义。氢键网络设计的困难之一是其形成需要多个位点的残基类型和侧链构象的协同变化。Boyken 等[55]在2016 年开发出一种计算方法HBNet 更充分地组合搜索残基类型和侧链构象,以快速枚举基于给定主链结构可能实现的所有侧链氢键网络。HBNet首先对所有极性侧链对应的所有构象(rotamer)之间的氢键和空间排斥相互作用进行预先计算。HBNet 的方法在2018 年得到了改进形成MC HBNet[56],使氢键网络的设计与计算速度更快。序列设计中保持主链结构固定对设计氢键网络有不利影响,未来可结合考虑主链柔性的设计技术来进行氢键网络设计。
2.4 负设计方法
蛋白质结构和功能并不直接取决于与单一结构状态对应的绝对自由能,而是取决于目标状态相对于其他状态的自由能差。例如,蛋白质折叠的稳定性取决于正确折叠态相对于非折叠态、错误折叠态、聚集态等的自由能差;分子间结合的亲和力取决于结合态相对于游离态的自由能差,等等。由于技术上的因素,绝大多数蛋白质计算设计仅考虑在目标结构状态下去优化氨基酸序列,以尽可能降低目标结构状态的自由能。这种聚焦于提高目标结构状态稳定性的设计思路被称为正设计(图7)。另一种可能的设计思路,则是提高目标状态之外其他结构状态的自由能,降低它们相对于目标结构的稳定性。这种思路被称为负设计(图7)。负设计机制被认为在天然蛋白质序列进化过程中普遍存在[57]。如果要在蛋白质设计中自动地考虑负设计,需要进行多状态设计,并引入目标状态之外的结构状态,通过改变序列使设计蛋白的目标结构和可能的竞争结构有明显的能量差距,这样设计出的氨基酸序列可以很容易地折叠为目标结构。而且仅仅关注目标结构并通过改变序列降低其能量有时可能不会改善目标蛋白质结构的折叠性,例如对于能量简并的竞争结构(蛋白质-蛋白质相互作用和螺旋低聚体)很容易产生的情况。所以需要考虑在降低目标结构能量的同时尽量提高其与其他状态结构的能量差距。
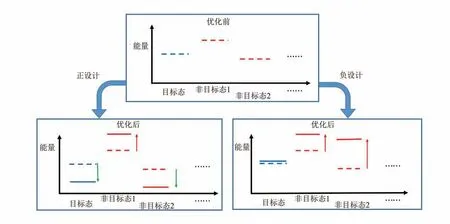
图7 正设计与负设计Fig.7 Positive design versus negative design
Hallen 等[38]在2017 年提出了一种多态蛋白质设计的通用程序,使用一个“适应度函数”来根据序列满足特定设计任务目标的程度来对多态蛋白质进行排名。通过首先将单个序列匹配到多个状态,计算该序列在每个状态上的能量,之后将这些能量合并以产生单个值,来评估适应度函数。通过每次迭代的多态设计,降低目标构象态的能量,扩大非目标态构象集的能量,最终达到多态设计的准确性。在2017 年他们又将多种多态设计方法与Rosetta 结合形成“Rosetta:MSF”,一种用于多状态计算蛋白质设计的模块化框架[58]。对有些问题,例如相互作用界面设计,基于多态设计引入负设计有一定的可行性。例如,为了增加蛋白质-蛋白质相互作用的特异性,可以利用负设计并惩罚那些有利于不良相互作用的序列。但是,需要考虑的蛋白质分子可能结构状态常常太多,这种显式考虑非目标状态进行负设计的方法至今没有较理想的策略,没有得到广泛应用。尽管如此,负设计作为一种概念和思想,仍然可以用来定性分析和比较不同的正设计结果。例如,全疏水的界面和亲/疏水组合的界面相比,后者可能功能上更优;实际上并非所有蛋白质设计任务都可以通过优化单个结构的序列来建模。
3 蛋白质计算设计应用
随着蛋白质计算设计技术的发展,在合成生物学、生物医学等领域逐步出现了相关的应用。本节中我们不区分从头设计和既有蛋白质改造,主要按设计目的不同分以下三类介绍不同的应用研究:通过蛋白质设计提高目标结构的稳定性;设计特异性的蛋白质小分子相互作用(包括酶活性中心);设计蛋白质分子间的特异性识别。
3.1 目标结构稳定性的提升
基于能量函数优化序列,常常获得结构稳定性非常高的设计蛋白。因此,蛋白质计算设计被用于指导蛋白质工程,改善天然蛋白的结构稳定性。另一个应用是疫苗设计,通过设计额外的主链骨架来维持抗原肽段的已知三维空间构象。此外,还可以通过序列重设计提高蛋白质在特定环境条件下的结构稳定性,如将膜蛋白改造为水溶性蛋白。以下将这几类应用分别举例说明。
Mu 等[3]利用Wijma 等[59]提出的FRESCO方案来提高酶稳定性。以黑曲霉葡萄糖氧化酶为突变对象,不仅根据FRESCO 方案,使用FoldX 和Rosetta_ddg 计算能量,还利用ABACUS进行能量计算,通过设定阈值来寻找远离活性位点的潜在稳定性突变位点。随后通过人工观察和分子动力学模拟来筛选突变集合,将提升稳定性的突变选项整合起来,最终得到多个稳定突变体。与野生型相比,突变体能够耐受更广泛的温度和pH 范围,并且显示出的催化活性更高,最好的突变体耐受温度较野生型提高了8.5 ℃,该突变体也在野生型会快速失活的pH6.0 和pH7.0 展现了更好的耐受性。Correia等[60]利用已知抗原结构来定义疫苗的功能构象,设计了稳定该构象的目标拓扑结构,而后用基于片段组装的方法从头设计出符合该拓扑结构的骨架,经过多轮的序列设计和主链优化的迭代,最终筛选出合理的结果并进行了实验验证。Marcandalli 等[61]通过设计蛋白质自组装纳米颗粒作为骨架来固定并呈递病毒性糖蛋白抗原复合物,从而在可控密度的条件下呈递此病毒抗原,实现疫苗的定制设计。Sesterhenn等[62]建立了TopoBuilder 系统,借此来从头设计能稳定复杂结构模体的蛋白质。通过这个系统,他们设计了能同时呈递三种抗原的蛋白,其设计方法为:针对不同的且结构复杂的抗原位点,首先在二维空间上列举适合的蛋白拓扑结构,并使用理想的二级结构单元和参数化设置将此二维空间投影至三维空间。通过这种方式,即可在不依赖模板的条件下设计所需的主链结构。
膜蛋白难以表达,且难以获得高分辨晶体结构,Slovic 等[63]对钾离子通道蛋白的表面进行位点突变设计,得到了其水溶性类似物。荧光蛋白的寡聚化是荧光蛋白应用的重要障碍,Wannier 等[64]通过表面突变设计和主链依赖的侧链rotamer 采样优化,得到了保持光强、不易聚集的红色荧光蛋白。
3.2 蛋白质-小分子相互作用的设计
通过重设计小分子结合界面,可以获得新的酶等催化元件、转录因子、荧光蛋白等化学感受元件。Banda-Vazquez等[65]通过口袋迁移(将一个天然口袋移植到另一个主链骨架上)和基于统计配对位置的搜索方法(获得与口袋残基突变关联但远离口袋的残基),对小分子结合蛋白LAOBP进行重设计,使其成为谷氨酰胺的结合蛋白。Glasgow等[66]参考了天然法尼基焦磷酸盐(FPP)-蛋白复合物模板,人工筛选了FPP 的结合口袋模体(仅包含4 个残基),而后通过与大量骨架界面对接、柔性骨架(骨架系综法)优化和序列设计的方法,设计了被FPP调节的生物效应器。为了设计能与高度缺电子的卟啉分子结合的非天然蛋白,Polizzi 等[67]通过数学参数化模型从头建立了反平行卷曲螺旋主链,并利用骨架系综法进行了柔性骨架设计。考虑到除了口袋位点以外,蛋白质核心区域的残基也可能会对其结合功能有影响,作者对所有内部残基和口袋位点进行了序列重设计,而非仅设计第一、二壳层的接触残基,最终设计出了高度热稳定的卟啉结合蛋白PS1。Dou等[68]在使用参数化方法首次成功从头设计β-桶蛋白的基础上,将其空腔与生色团3,5-二氟-4-羟基亚苄基-咪唑啉酮进行对接设计,得到了从头设计的荧光蛋白。Li等[4]通过底物结合口袋的重设计,将芽孢杆菌YM55-1 天冬氨酸酶狭窄的催化底物范围拓展到作为互补氢胺化反应,且对底物耐受性最高达到300g/L,定向改变了芽孢杆菌YM55-1天冬氨酸酶的催化功能。
3.3 蛋白分子间的特异性识别设计
Leaver-Fay 等[69]、Froning[70]提出了设计双特异性抗体的方法,使用多状态设计策略,并考虑引入非目标状态进行负设计。Silva 等[71]从头设计了一个有着天然细胞因子结合位点,然而拓扑结构和序列都不同于天然蛋白的人工细胞因子,此设计蛋白只结合天然白细胞介素-2的部分受体,却不结合其他受体,隔绝了对部分下游细胞信号的影响。Chen 等[72]通过用参数化的方法从头设计螺旋主链骨架,并建立氢键网络、环区的连接,进行序列优化,获得多组具有特异性异源二聚能力的蛋白对,并用它们构建了蛋白质逻辑门[73]。Langan 等[74]针对信号通路中天然存在的相互作用蛋白,将控制蛋白功能的“笼子”、“插销”和“钥匙”分别设计于蛋白相互作用界面上,通过界面设计实现了调节某对蛋白相互作用的人工蛋白开关设计,并把这一设计用于内源性信号通路的反馈控制[75]。
蛋白质分子间的特异性识别设计也牵涉到组装体的设计,其可以应用于新材料领域。Shen等[76]进行了蛋白质自组装体的从头设计,使其可以聚集成微米级的细丝。他们首先建立了一个纤维片段,随后通过旋转平移形成参数化的螺旋结构,再在这个骨架基础上进行序列设计。根据纤维片段和旋转平移等参数的变化,可以形成大量不同的蛋白,这一设计策略有助于推动一系列多尺度超材料的制造。King等[77]更新了Rosetta的对称建模框架tcdock,用来模拟高度有序对称的蛋白质支架对的对接,依据每一个对接构型对界面设计的实用性打分,最后使用负染电镜等手段对设计出的蛋白质的组装状态进行X 射线晶体学分析,结果表明设计出的组装体材料与理论值的RMSD偏差在0.5~1.2 Å,证明了这种方法对界面几何形状有着精确的控制,并且能够高精度地设计具有多种纳米级特征的双组分蛋白质纳米材料。Fallas等[78]采用类似于软质心模型的Monte Carlo Sampling,首先生成用于对接的主链模型,然后使用骨架原子的坐标和二级结构元件来对蛋白质-蛋白质对接进行打分,最后使用全原子Rosetta Design[25]计算优化蛋白质-蛋白质界面序列,结果表明所设计的蛋白质在溶液中稳定地形成均聚物。
4 展 望
蛋白质计算设计把我们对蛋白质序列-结构-功能关系的生物物理认识和数学模型、计算方法等综合在一起,逐渐形成了一套系统的理论和方法学,并得到越来越多的实验验证,展示出广泛应用前景,是合成生物学的重要使能技术之一。
蛋白质计算设计的发展和应用仍然处于初级阶段。从方法上来讲,主链结构和功能的从头设计的效果还有很大提升空间。已有的关于主链的设计方法,一般是基于天然片段进行主链设计,亦或是对规则结构进行参数化设计。而当前的能量函数还不能完全做到主链的全自动从头设计,对极性相互作用的定量刻画还不够准确。基于rotamer 的构象表示方法为极性相互作用的定量化带来困难:离散rotamer 对侧链原子位置引入较大误差,不能准确地进行氢键网络设计。未来方法进一步发展的关键包括对主链设计能量模型和侧链极性相互作用模型的改进。
由于蛋白质并非孤立存在的,这既体现为蛋白质的功能往往与其他生物分子(如磷脂双分子层)互作有关,也体现为细胞内外环境(如pH)为蛋白质提供的复杂溶剂环境。而目前的设计方法中,往往是将其他小分子视做刚体进行对接,并将蛋白质周围环境进行简化估计。尽管这些简化模型是出于对效率的考量,它们在实际应用中对成功率的影响也是不能忽视的。目前已有关于将pH[79-80]、磷脂双分子层[22,81-83]等方面因素引入蛋白质设计的分析。这些尝试有望拓宽蛋白质计算设计方法的应用领域,也有望提高蛋白质设计的合理性和成功率。此外,如何在蛋白质计算设计的整体框架中考虑和处理负设计,也是未来方法研究的要点之一。
同时,由于蛋白质-蛋白质界面的形状和化学特征的极端多样性,确定蛋白质的识别位点和能量热点的简单策略一般不会很有效[84]。因此,建立起氢键和静电相互作用易于计算的描述,对于蛋白质-蛋白质界面的能量函数的充分建模非常重要。一个相关的挑战是建立合理的水分子模型,这些水分子通常在蛋白质界面上形成水介导氢键的延伸网络,而标准的隐式溶剂化模型无法捕捉到这些网络[85]。除了能量函数,在主链柔韧性的建模方面也存在缺陷。解决这些问题对于基于结构的蛋白质相互作用特异性的深刻理解和预测至关重要。因此还需要通过更精确的建模技术生成详细和精确的结构模型来模拟界面[85]。
随着方法成熟度提高,蛋白质计算设计将越来越多地被应用于功能蛋白设计,这包括各类蛋白质探针和传感器的设计和改造以及酶的设计。比如在医学中,抗体等诊断和治疗蛋白的设计、疫苗设计等;在合成生物学中,酶等催化元件以及感受器、逻辑门等人工调控元件的设计也将会成为蛋白质计算设计的重要应用领域。
猜你喜欢
生物化学与生物物理进展(2022年7期)2022-07-25
生物化学与生物物理进展(2022年6期)2022-07-21
物理学报(2022年10期)2022-06-04
西南农业学报(2021年10期)2021-12-14
山东农业大学学报(自然科学版)(2021年3期)2021-07-29
中学生数理化(高中版.高考理化)(2021年2期)2021-03-19
世界农药(2019年2期)2019-07-13
水运管理(2018年5期)2018-08-20
教育教学论坛(2017年14期)2017-04-20
湖北农业科学(2012年16期)2012-09-12
