
中枢神经系统原发性黑色素细胞瘤2例临床病理分析
2021-03-17 05:58蔡丽君王代忠蔡宇翔汪必成田素芳
临床与实验病理学杂志 2021年1期
蔡丽君,王代忠,蔡宇翔,汪必成,田素芳
中枢神经系统(central nervous system, CNS)原发性黑色素细胞瘤(primary meningeal melanocytoma, PMM),即脑脊膜黑色素细胞瘤是一种罕见的交界性病变,其发病率约为1/1 000万,占脑肿瘤的0.06%~0.1%[1]。CNS PMM好发于后颅窝、桥小脑角、Meckel腔及颈段脊髓的髓外硬膜腔内,表现为境界清楚的实性肿块。组织学上,肿瘤细胞呈漩涡状或片状分布,细胞形态相对温和,细胞质内含多少不等的黑色素颗粒。该肿瘤虽然缺乏间变特征,但术后复发率较高,也可累及脑脊髓组织。CNS PMM需与其它含有色素的肿瘤鉴别,由于病例罕见,相关文献多为个案报道,缺乏对该肿瘤临床病理资料的积累。本文现报道2例CNS PMM,探讨其临床病理学特征、免疫表型、诊断及鉴别诊断,并复习相关文献,旨在提高病理医师对该病的认识水平。
1 材料与方法
1.1 材料收集武汉大学中南医院及湖北省十堰市太和医院2013~2019年诊治的2例CNS PMM,回顾性分析其临床病理资料。临床信息及影像学资料来自电子病历系统,随访信息来自患者的电话回访。
1.2 方法手术标本均经10%中性福尔马林固定,常规脱水、石蜡包埋。4 μm厚切片,行HE染色及免疫组化检测,免疫组化染色采用EnVision两步法,所用一抗HMB-45、S-100、Melan-A、EMA、PR、GFAP、Olig-2、SSTR2及Ki-67均购自北京中杉金桥生物公司,常规设阳性和阴性对照。例1因肿瘤组织黑色素颗粒含量较高,切片经抗原修复后,置于PBS(pH 7.4~7.6)配制的浓度为1.8%的H2O2溶液中70 ℃水浴箱孵育2 h[2],镜下观察完全脱去黑色素后进行后续免疫组化检测。
2 结果
2.1 临床特点例1女性,42岁,因右侧前额及颞部麻木疼痛伴右眼视力下降7天入武汉大学中南医院。MRI示右侧海绵窦旁-颞极部见混杂短T1(图1A)短T2(图1B)低FLAIR信号肿块,大小24 mm×13 mm×20 mm,增强呈明显欠均匀强化,经眶下裂累及翼腭窝,与邻近脑膜宽基底相连,考虑脑膜黑色素瘤可能,遂行肿瘤切除。术后第4天行头颅MRI复查,显示肿瘤部分切除。例2男性,初次发病22岁,2003年于湖北省十堰市太和医院行第2~3颈椎椎管内硬膜下肿瘤切除,病理诊断为PMM;2013年因发现同一部位肿瘤,考虑为PMM复发,遂再次行手术切除。
2.2 病理特征例1送检大小2 cm×1 cm×0.5 cm灰红、灰褐色不整形碎组织一堆。镜下肿瘤与硬脑膜相连,呈漩涡状或片状分布(图2A),肿瘤细胞呈梭形、多边形或圆形,其内含大量黑色素颗粒,细胞无明显异型性,未见明显核分裂象(图2B),未见明确出血及坏死。例2于2003年和2013年的切片均显示肿瘤组织与硬脊膜相连,呈片状、巢团状或围绕血管呈乳头状分布,肿瘤细胞呈梭形或圆形,部分细胞胞质内含黑色素颗粒,细胞核呈圆形、豆状或空泡状,部分细胞可见核沟及小的嗜酸性核仁,核多形性及核分裂象不明显(图3A)。
2.3 免疫表型例1切片脱黑色素后免疫组化染色显示HMB-45、Melan-A均阳性,EMA、PR、GFAP、Olig-2及SSTR2均阴性,Ki-67增殖指数约1%。例2的两次手术病理切片免疫组化染色均显示HMB-45(图3B)、Melan-A及S-100均阳性,EMA及PR均阴性,Ki-67增殖指数约1%。

图1 例1 MRI检测:A.右侧海绵窦旁-颞极部肿块T1WI高信号,边界尚清;B.肿块T2WI呈低信号
2.4 治疗及随访例1患者行肿瘤局部切除术后辅以放疗,术后半年随访,未见复发。例2患者2013年第2次肿瘤切除术后随访至今,未见再次复发。
3 讨论
CNS原发性黑色素细胞性肿瘤是一类原发于CNS的黑色素肿瘤,起源于软脑膜黑色素细胞,该细胞由神经嵴衍生而来[3]。在CNS中,黑色素细胞在正常情况下主要分布于大脑基底部、延髓腹侧和颈脊髓上段。CNS原发性黑色素细胞性肿瘤临床较罕见,根据WHO CNS肿瘤分类[1],其主要分为4类。(1)良性弥漫性病变,未形成肉眼可见的肿块:黑色素细胞增多症(melanocytosis);(2)恶性弥漫性或多灶性病变:黑色素瘤病(melanomatosis);(3)良性或中间性肿瘤:黑色素细胞瘤(melanocytoma);(4)恶性孤立性肿瘤:黑色素瘤(melanoma)。黑色素细胞增多症及黑色素瘤病与神经皮肤黑变病(一种罕见的斑痣性错构瘤病,多发生于2岁前,主要表现为先天性巨大或多发的皮肤黑色素痣伴CNS黑色素细胞增生)密切相关,可能起源于黑色素细胞前体细胞,它们在获得体细胞突变(主要是NRAS)后迁移至CNS[4]。
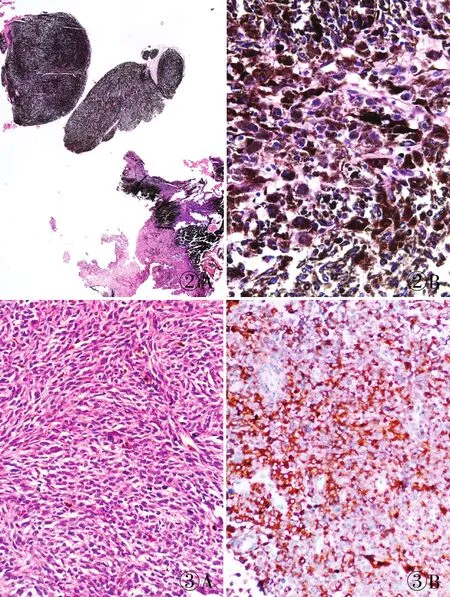
图2 例1:A.肿瘤与硬脑膜相连,呈漩涡状或片状分布;B.肿瘤细胞内含大量黑色素颗粒,细胞无明显异型性,未见明显核分裂象 图3 例2:A.肿瘤细胞呈梭形,片状分布,核多形性及核分裂象不明显;B.肿瘤细胞HMB-45阳性,EnVision两步法
CNS PMM可发生于任何年龄(9~73岁),最常见于45~50岁,男女比为1 ∶1.5;多单发,个别多发[5],结节状,呈棕黑色或无色素,不侵犯脑实质和脊髓。本组例1和例2,初次发病年龄分别为42和22岁,均为单发肿块,未侵犯周围神经组织。PMM在MRI检查下可因黑色素的顺磁性而呈现特征性表现,在T1加权像上呈等或高信号,在T2加权像上呈低信号,增强呈均匀或不均匀强化。组织学上,肿瘤细胞呈漩涡状、巢状、片状或乳头状排列,细胞相对温和,呈上皮样、梭形或圆形,胞质内含黑色素颗粒,细胞核呈卵圆形、豆状或空泡状,可有核沟及小的嗜酸性核仁,核多形性及非典型性不明显;无出血、坏死及CNS侵犯,核分裂象<1个/10 HPF。若侵犯CNS或核分裂象增加,则划分为中间级别黑色素细胞瘤[6]。黑色素细胞瘤免疫表型:vimentin、S-100、HMB-45、Melan-A均阳性,S-100敏感性高但特异性不高,SOX10、Tyrosinase及MITF亦阳性,GFAP、CK、EMA均阴性,Ki-67增殖指数<2%;Ⅳ型胶原和网状纤维染色显示血管周及肿瘤细胞巢周围网状纤维包绕,但在单个细胞周围无网状纤维[1]。电镜下胞质内有不同成熟阶段的黑色素小体及前黑色素小体,有少数中间连接,无桥粒和连接复合体。
近年来研究发现[4,7],CNS原发性黑色素细胞性肿瘤常存在GNAQ、GNA11及NRAS等热点突变,其中GNAQ和GNA11突变多见于成人,NRAS突变多见于儿童。GNAQ或GNA11是Gq家族中两个密切相关的G蛋白,它们编码蛋白质GTPase功能所需的关键氨基酸[8]。GNAQ和GNA11作为起主导作用的致癌基因,激活包括MAP-kinase通路在内的多个关键信号通路,进而导致肿瘤性病变[9]。
CNS PMM首先需与CNS原发性黑色素瘤鉴别:后者属于高度恶性肿瘤,为孤立性肿块,呈侵袭性生长,临床进展迅速,预后差,可转移至远处器官。CNS原发性黑色素瘤与硬膜相连,可发生于神经轴的任何位置,以脊髓和后颅窝居多[10]。组织学上,与PMM相比,CNS原发性黑色素瘤核异型性、核间变更易见,可见怪异核,细胞密度更大,核分裂象更多,可见出血、坏死及明确的组织侵犯[1]。Ki-67增殖指数常超过8%[6]。CNS原发性黑色素瘤可有GNAQ或GNA11突变(频率低于PMM)、NRAS突变及BAP1失活等。
除此之外,CNS PMM的诊断还需充分结合临床、影像学及组织学特征,与其它黑色素性肿瘤鉴别:(1)转移性黑色素瘤,多位于脑实质内且多发,有原发病史,组织学上常见多形性及坏死。TERT启动子、NRAS、BRAF和KIT基因突变多见于皮肤和肢端黑色素瘤,这些突变可能有助于提示为转移灶。但值得注意的是,大多数儿童CNS原发性黑色素细胞性肿瘤,尤其是与神经皮肤黑色素病相关的黑色素肿瘤也是由NRAS突变驱动的[1]。(2)脑膜瘤,可含有内陷的脑膜黑色素细胞,形态学可与PMM相似;但免疫组化标记SSTR2、EMA、PR、vimentin均阳性,HMB-45等黑色素标记阴性。电镜检查可见特殊的细胞间连接及桥粒,细胞周围有基板。目前WHO认为无“黑色素性脑膜瘤”之称,之前称为黑色素脑膜瘤应属于黑色素细胞瘤[1]。(3)色素性神经鞘瘤,是一种能产生黑色素的少见神经鞘瘤亚型,好发于中枢神经根和交感神经节。镜下部分瘤细胞内含有多少不等的黑色素颗粒,免疫表型与黑色素细胞瘤有重叠,鉴别诊断主要靠形态学观察及电镜观察。电镜下瘤细胞周围有广泛的基膜样物质沉着。有研究表明,GNAQ突变对CNS原发性黑色素细胞性肿瘤与色素性神经鞘瘤的鉴别诊断有重要意义,与色素性神经鞘瘤相比,GNAQ突变在CNS原发性黑色素细胞性肿瘤中的特异性可达100%[11]。(4)伴色素的室管膜瘤,瘤细胞密度中等,形态一致,可见围绕血管的假菊形团,免疫组化标记GFAP、S-100、vimentin和EMA均阳性,HMB-45和MART-1均阴性[12]。
CNS PMM治疗以手术切除为主,由于肿瘤过度出血、变硬或附着于正常垂体和鞍膈的蛛网膜,完整切除常不易实现[13]。局部切除后可辅以放疗,肿瘤完整切除患者预后明显优于局部切除患者[14]。CNS PMM虽为交界性肿瘤,但术后复发率较高。有学者回顾性分析文献报道的95例脑膜PMM,其复发率达26.3%,与肿瘤相关的病死率为10.5%[15]。本组例2为肿瘤切除10年后复发;例1术后半年随访,预后良好,未见复发。因此患者术后应注意密切随访,定期复查。
猜你喜欢
养生月刊(2022年4期)2022-11-26
林业科技(2022年4期)2022-08-06
昆明医科大学学报(2022年8期)2022-07-31
医学研究杂志(2022年3期)2022-03-29
昆明医科大学学报(2021年12期)2021-12-30
昆明医科大学学报(2021年4期)2021-07-23
实用肿瘤学杂志(2020年6期)2020-12-09
阅读与作文(小学低年级版)(2019年12期)2019-12-26
奥秘(创新大赛)(2019年8期)2019-11-30
小天使·四年级语数英综合(2019年9期)2019-11-09
