
不同来源的大豆水溶性多糖乳状液乳化特性及红外光谱分析
2021-03-17 10:34李佳怡郭思晗崔怀田卢苗苗
渤海大学学报(自然科学版) 2021年4期
李 君,李佳怡,郭思晗,齐 妍,崔怀田,卢苗苗,宋 虹,刘 贺
(渤海大学 食品科学与工程学院,辽宁 锦州 121013)
0 引言
大豆副产物高值化利用近年来备受关注,大部分豆渣、豆皮被用作饲料、肥料或废弃物丢弃处理,造成了资源的浪费和环境的污染[1].因此大豆副产物的综合利用还需要进一步的探索和实践.大豆多糖属于水溶性植物多糖,除了具有吸水能力强、粘度高、在溶液中分散性高等特性外,其凝胶性、乳化性以及增稠性也较好.因而大豆多糖作为食品添加剂被广泛用于替代食物脂肪组织中,并且在提高食品组织状态稳定性和开发功能性食品等方面也多有应用.另一方面,大豆多糖作为一种膳食纤维,可以通过降低胃肠道的消化吸收率来增加饱腹感,并且在肠道的消化过程中,以发酵纤维作为益生元,来增加乳酸杆菌以及双歧杆菌的数量,从而达到降血脂、降血压、降血糖的目的[2-4].多糖和蛋白质作为乳化剂在食品中应用,其效果并不理想,若用多糖和蛋白质共聚物来稳定乳液,其效果由蛋白质与多糖的作用性质以及自身性质来决定[5].以往大豆多糖以酶法、离子交换树脂法和酸法提取为主,近年来,微波和超声波等辅助提取大豆多糖的方法得到广泛应用,且在该领域取得了较大的成就.
目前大豆多糖的研究集中在单一多糖的乳化性质对乳状液的影响,没有进行同种来源的不同副产物中大豆多糖的乳化性质对乳状液影响的横向比较.因此,本试验利用微波辅助草酸铵提取四种大豆多糖,测定傅立叶变换红外光谱、乳状液的界面张力、粒度分布、Zeta电位、光学显微镜、流变性质、多重光散射等指标,探讨不同副产物中提取的大豆多糖-蛋白乳状液乳化特性的影响规律,为未来进一步对大豆副产物的综合利用研究奠定基础.
1 材料与方法
1.1 原料与试剂
山东禹王集团:大豆种皮、全豆豆渣、脱蛋白豆渣、子叶豆渣.
乙醇:天津市天力化学试剂有限公司;草酸铵:天津市大茂化学试剂厂.
1.2 仪器与设备
LAB多重光散射仪:Turbiscan,北京郎迪森科技有限公司;视频光学接触角测量仪:OCA 15EC,德国dataphysics公司;傅立叶变换红外光谱仪:Scitar2000,原瓦里安;激光粒度分布仪:NANO ZS90,英国马尔文仪器有限公司;流变仪:DHR-1,美国TA公司;光学显微镜:尼康80i,北京瑞科中仪科技有限公司;数显高速分散均质机:FJ200-SH,上海标本模型厂.
1.3 试验方法
1.3.1 大豆多糖的制备
将大豆种皮、子叶豆渣、全豆豆渣、脱蛋白豆渣置入鼓风干燥箱中烘干,用锤式旋风磨粉碎,过60目筛,每100 g原料加入1%的乙醇溶液10 mL进行脱色,25 ℃下持续搅拌30 min后,利用双层纱布过滤,将残渣放置于鼓风干燥箱中在65 ℃下进行烘干.之后再精确称量烘干后的原料粉末50 g,加入6 g的草酸铵,微波90 ℃温度下,每次微波5 min,重复操作4次,双层纱布进行过滤,上清液在3 500 r/min的条件下离心为15 min,后经旋转蒸发,再浓缩至上清液体积的1/3,用0.1 mol/L的盐酸溶液调节pH至4.0,缓慢的加入双倍浓缩液质量的无水乙醇并不断搅拌,在4 ℃条件下静置12 h,进行双层纱布过滤,所得物放在65 ℃恒温干燥箱中烘干,得到四种大豆粗多糖.
1.3.2 乳状液制备方法
将1 g大豆多糖溶解于50 mL水中形成多糖水溶液.随后将10 mL大豆油与40 mL水进行混合,再利用高速剪切机在10 000 r/min条件下剪切1 min,得到初级乳状液.多糖水溶液50 mL与初级乳状液50 mL进行均匀混合,再利用高速剪切机在10 000 r/min的条件下剪切1 min,得到次级乳状液.最后利用高压均质机50 MPa压力下循环均质6 min,最终得到100 mL多糖乳状液,再添加0.02%的叠氮化钠确保乳状液在长时间储存下不会变质.制成的乳状液放入冰箱在4 ℃下进行低温储存.
1.3.3 乳状液界面张力测定
利用视频光学接触角测量仪测定界面张力,试验在25 ℃的常温下进行,使用移液枪移取1 mL的乳状液,并通过测量其质量得到密度值,再将乳状液置于注射器内,挤出7 mL的样品溶液通过校准后,测量三次平行并记录数据,可以得到界面张力[6].
1.3.4 乳状液粒径分布测定
采用BT_9300ST激光粒度分布仪来测定乳状液的粒度分布情况,同时记录中位径、表面积分数平均粒径(D3,2)及体积分数平均粒径(D4,3)[7].参数设定为:散射角度90°,激光波长633 nm,温度25 ℃,颗粒折射率1.596,颗粒吸收率0.001;分散剂设定为水,分散剂折射率1.333.每个样品测量三次,结果取平均值.
1.3.5 乳状液Zeta电位测定
通过NANO ZS90激光粒度分布仪来测定乳状液的Zeta电位,吸取1 mL稀释至10倍的乳状液于样品池中,在20 ℃的温度下进行Zeta电位测定[8].每个样品测量三次,结果取平均值.
1.3.6 乳状液光学显微镜测定
稀释10倍的乳状液充分混匀后,滴于载玻片上,待其均匀铺展开后将盖玻片的一侧放置到液滴的边缘,轻轻地将盖玻片覆盖于乳状液液滴表面,注意避免气泡产生,再将载玻片放置于光学显微镜下(40倍物镜×10倍目镜),观测乳状液的形态.
1.3.7 乳状液流变性质测定
利用DHR-1流变仪测定乳状液的流变性质,取适量乳状液置于测试台上,在25 ℃条件下,使用40 mm平行板夹具,狭缝距离设定为0.5 mm,剪切速率为0~100 s-1,检测样品的流变特性[9],每个样品黏度重复测量三次.
1.3.8 傅立叶变换红外光谱(FT-IR)测定
采用压片法制样,样品除去游离水后,称量1 mg与KBr进行混合研磨,制成压片后.在样品测定之前,通过背景扫描除去干扰因素,在中红外范围:400~4 000 cm-1区内进行红外光谱扫描[10].
1.3.9 乳状液多重光散射测定
采用Turbiscan LAB多重光散射仪对乳状液进行扫描,可以得到随时间延长产生的不同响应值的曲线[11].对乳状液进行扫描,间隔1 min,共扫描20 min,进行TSI对比,TSI的最终数值的大小和曲线斜率变化会反映乳状液状态是否稳定.
1.4 数据统计及分析
本试验采用Origin 2017和Excel 2010进行绘图和制表,对界面张力、粒度分布、Zeta电位、流变性质、傅立叶变换红外光谱、多重光散射进行数据分析.
2 结果与讨论
2.1 乳状液界面张力分析
本试验采用悬滴法测定乳状液的界面张力如图1.大豆种皮多糖乳状液界面张力最小为51.15±0.17 mN/m,全豆豆渣多糖乳状液界面张力为51.87±0.25 mN/m,脱蛋白豆渣多糖乳状液和子叶豆渣多糖乳状液界面张力分别为52.76±0.38 mN/m和53.28±0.06 mN/m.大豆种皮多糖乳状液和全豆豆渣多糖乳状液的界面张力小,具有更好的界面活性,这可能是大豆种皮多糖乳状液和全豆豆渣多糖乳状液中多糖的支链较多,在油水界面上吸附更多的蛋白质分子,蛋白质和多糖交联形成层层沉积的界面膜结构,导致界面张力较小,从而提高乳浊液的稳定性.这与于培玲等[12]的研究结果一致.Peter等[7]研究发现在pH 3.0下提取出的豆渣多糖比pH4.0~5.0下提取出的豆渣多糖更能显著降低界面张力,可能是因为其蛋白含量较高,或与其多糖构象有关.

图1 乳状液界面张力
2.2 乳状液粒径分布分析
乳状液粒度大小及其分布是决定乳状液是否稳定的重要参数[13],液滴分布越集中,粒径越小,则乳状液越趋于稳定.由表1可以看出,大豆种皮多糖乳状液和全豆豆渣多糖乳状液的体积平均径和面积平均径要远远小于脱蛋白豆渣多糖乳状液和子叶豆渣多糖乳状液的体积平均径和面积平均径.图2为乳状液的粒径分布图.由图2可知,大豆种皮多糖乳状液和全豆豆渣多糖乳状液的粒度分布更加集中,粒径更小,峰值更高,而乳状液也更为粘稠,更加稳定.而脱蛋白豆渣多糖乳状液和子叶豆渣多糖乳状液的粒度分布较为宽泛,并且粒度分布曲线向粒度较大方向偏移,导致乳状液的分层较快,其稳定性也更差.这可能是由于大豆种皮多糖乳状液和全豆豆渣多糖乳状液中蛋白质分子在油水界面的吸附量较大,蛋白质分子间静电排斥和空间排斥作用使乳状液粒径减小,降低油滴聚集,从而提高乳状液稳定性[14].这与李超[15]对紫苏蛋白O/W乳状液的粒径分布及大小研究中的乳状液粒径减小,降低油脂聚集,能够提高乳状液稳定性这一结论相一致.

表1 乳状液粒径参数

图2 乳状液粒径分布图
2.3 乳状液Zeta 电位分析
乳状液表面的界面电荷会影响乳液的稳定性.Zeta电位是表明分散体系稳定与否的重要指标,该指标决定了乳状液在外界环境中是否稳定.Zeta电位的测定可以预测乳状液是否出现凝聚和絮状凝结的情况.Zeta电位的绝对值越大,液滴间的就斥力越大,乳状液就越不容易聚结,因此也就越稳定.由表2可知,大豆种皮多糖乳状液和全豆豆渣多糖乳状液的Zeta电位的绝对值更大,表示乳状液稳定性较好,而脱蛋白豆渣多糖乳状液和子叶豆渣多糖乳状液的Zeta电位的绝对值更小.这说明大豆种皮多糖乳状液和全豆豆渣多糖乳状液中蛋白质在水相和油相之间空间位阻更大,液滴之间更不容易形成吸附,乳状液也更加稳定,更不容易聚集[16].Zeta电位与上述界面张力和粒径分布结果一致.

表2 乳状液Zeta 电位
2.4 乳状液流变性质分析
图3为剪切速率升高对乳状液黏度的变化情况.由图可知,随着剪切速率的增加,大豆种皮多糖乳状液的黏度最高,这说明乳状液液滴表面的界面电荷之间的吸附能力更强,蛋白质能更好的吸附在乳状液的油滴表面,剪切后还能保持较好的吸附状态,保持乳状液的黏度.而全豆豆渣多糖乳状液、脱蛋白豆渣多糖乳状液和子叶豆渣多糖乳状液的黏度则较低,这说明液滴表面的电荷发生了分散,电荷之间的吸附率降低,液滴表面的蛋白质吸附量降低,导致乳状液的黏度也随之降低.图4为剪切速率对乳状液剪切应力的影响.由图可知,随着剪切速率的加快,乳状液体系的总体剪切应力从较低程度向较高程度持续发展.其中添加大豆种皮多糖的乳状液的剪切应力最大,这可能是由于液滴表面的电荷吸附力较强,使其分散需要对其做功更多.而添加全豆豆渣多糖、脱蛋白豆渣多糖、子叶豆渣多糖的乳状液液滴表面电荷被打散,导致电荷间的吸附能力降低,使乳状液分散所需做功较少,表现出乳状液的剪切应力较小.
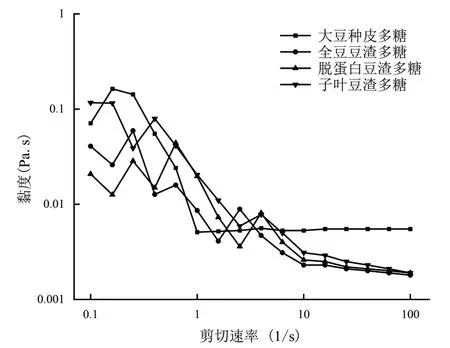
图3 剪切速率对黏度的影响

图4 剪切速率对剪切应力的影响
2.5 乳状液多重光散射分析
乳状液的稳定性可以通过TSI稳定系数的变化来体现,TSI稳定系数最终值越小,斜率越小,说明随着时间延长,乳状液的变化越小,乳状液越稳定[17].
如图5所示,经过20 min后,大豆种皮多糖乳状液和全豆豆渣多糖乳状液的TSI达到0.8左右,明显小于脱蛋白豆渣多糖乳状液和子叶豆渣多糖乳状液的TSI,其值达到1.3左右,并且大豆种皮多糖乳状液和全豆豆渣多糖乳状液的曲线斜率也明显小于脱蛋白豆渣多糖乳状液和子叶豆渣多糖乳状液的曲线斜率.这说明经过相同的时间,大豆种皮多糖乳状液和全豆豆渣多糖乳状液的TSI稳定系数最终值更小,斜率也更小,其稳定性也更好[18].这可能是大豆种皮多糖和全豆豆渣多糖的支链较多,更易于形成油水界面的蛋白质和多糖的复合物,再通过扩散和对流,多糖逐渐被吸附到界面上,开始展开过程以及疏水基团的暴露,从而增加了界面活性,导致稳定性的提高[19].并且从TSI曲线斜率的增长趋势来看,相比于添加大豆种皮多糖的乳状液,添加全豆豆渣多糖的乳状液的曲线斜率更加趋近于平稳,说明该乳状液逐渐趋于稳定,稳定性更好.
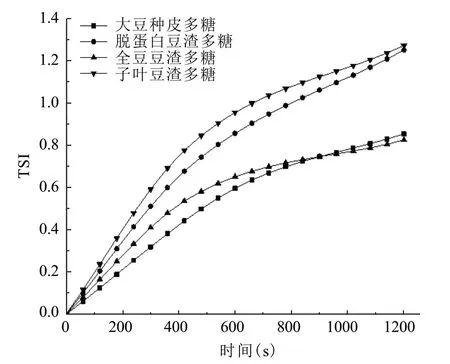
图5 TSI随时间变化曲线
2.6 乳状液光学显微镜分析
图6为乳状液光学显微镜观测图.由图可知,大豆种皮多糖乳状液和全豆豆渣多糖乳状液的颗粒较小且更为均匀,没有明显的颗粒聚集情况出现.其中大豆种皮多糖乳状液的颗粒更为密集,这表明该乳状液具有更多的蛋白质和多糖交联形成层层沉积的界面膜结构,黏度更高并且更加均匀不易分层.而全豆豆渣多糖乳状液虽然也具有很好的稳定性,但乳状液黏度较之略低;脱蛋白豆渣多糖乳状液和子叶豆渣多糖乳状液的颗粒较大且更为分散,并且在图片中可以看出明显的乳状液颗粒的聚集,这表明该乳状液中的蛋白质和多糖复合物较少,更易出现相分离,发生分层现象,并且乳状液的黏度也更低,这与Zeta电位分析的结果相一致.并且也与乳状液粒度分布的分析中,大豆种皮多糖乳状液和全豆豆渣多糖乳状液的粒径小于脱蛋白豆渣多糖乳状液和子叶豆渣多糖乳状液的粒径这一结果相互印证.

图6 乳状液光学显微镜观测图(400×)
2.7 傅立叶变换红外光谱(FT-IR)分析
图7为大豆种皮多糖、全豆豆渣多糖、脱蛋白豆渣多糖和子叶豆渣多糖的傅立叶变换红外分析图谱,检测多糖所含有的官能团.不同的官能团会在分析图谱中体现在不同的特征峰上,通过对特征峰的比对,可以比较出不同种多糖之间存在的官能团差异,并且由此可以对不同大豆多糖的乳化性质的差异有所了解[20].

图7 傅立叶变换红外光谱分析图
由图7可知,大豆种皮多糖和全豆豆渣多糖的特征吸收峰相似程度较高.大豆种皮多糖和全豆豆渣多糖红外图谱中3 500~3 000 cm-1的两个宽峰是O-H和N-H的伸缩振动[21],由于多糖分子间或分子内的羟基发生缔合,2 923 cm-1附近的吸收峰由C-H的伸缩振动所引起[22],位于1 650~1 500 cm-1的特征峰是蛋白质的二级结构β折叠所引起的N-H的变角振动[23],这说明含有羟基和氨基,1 438 cm-1左右的峰是由C-O伸缩振动引起[24],769 cm-1的峰是吡喃环对称环伸缩振动引起[2].由此可以说明两种粗多糖中多糖与蛋白结合,形成糖蛋白.尽管两种多糖具有很多相似的特征吸收,但还是略有差异.大豆种皮多糖红外图谱中1 147 cm-1左右的峰是由吡喃环的醚键引起,而全豆豆渣多糖红外分析图谱中1 735 cm-1的峰是由GalA羧基形成的酯键C═O的伸缩振动引起[25].
与大豆种皮多糖相比,脱蛋白豆渣多糖红外分析图谱中没有位于1 650~1 500 cm-1的特征峰,也就没有蛋白质的二级结构β折叠所引起的N-H的变角振动,而子叶豆渣多糖红外分析图谱中没有由O-H和N-H的伸缩振动引起的位于3 500~3 000 cm-1的两个宽峰和由C-O伸缩振动引起的位于1 438 cm-1左右的吸收峰,由此可以得出结论大豆种皮多糖的乳化性质优于脱蛋白豆渣多糖和子叶豆渣多糖.
3 结论
本研究从不同大豆副产物中提取大豆水溶性多糖,通过测定其乳状液的界面张力、粒度分布、Zeta电位、多重光散射、流变性质、微观结构及傅立叶变换红外光谱对乳状液的乳化特性进行比较分析.结果表明,四种大豆多糖的乳化能力从强到弱为:大豆种皮多糖、全豆豆渣多糖、脱蛋白豆渣多糖、子叶豆渣多糖.大豆种皮多糖乳状液的界面张力最低(51.15 mN/m),Zeta电位绝对值最高(-24.9 mV),剪切应力和黏度也最高,粒度分布更加集中且均匀,粒径更小,TSI稳定系数更小,斜率更小,表明大豆种皮多糖的乳化能力最强,乳状液的稳定性最好.本研究结果不仅为未来研发大豆种皮多糖制品提供了一定的理论基础,还可促进对食品新资源的重新利用,减少对资源的浪费.
猜你喜欢
粮油与饲料科技(2022年2期)2022-11-24
粮食科技与经济(2020年2期)2020-05-09
祝您健康·文摘版(2019年12期)2019-12-13
学生导报·东方少年(2019年3期)2019-05-14
湖北农业科学(2017年6期)2017-04-26
凤凰生活(2016年3期)2016-03-08
江苏农业科学(2015年11期)2016-01-27
长江蔬菜·学术版(2014年7期)2015-01-21
农家顾问(2014年4期)2014-06-26
