
水热驱动的Claisen重排-亲核加成串联反应法合成3-甲基-5-羟基-7-羧基异色满-1-酮
2021-03-13 08:32张广威胡海慧李双双卢文欣
合成化学 2021年2期
张广威,胡海慧,李双双,卢文欣,王 鹏
(山东科技大学 化学与生物工程学院,山东 青岛 266590)
异色满酮所具有的苯并六元环内酯结构是常见的有生物活性的结构,常见于各类药物的结构中[1-3],是一种重要的药物中间体。合成异色满酮常用的方法主要是苯并环类化合物的氧化[4]、羰基化合物的环加成[5]、芳香醇类化合物的羰基化反应[6]、金属催化反应[7]等,但这些方法或多或少都存在反应条件苛刻、原料结构复杂、反应步骤繁复等缺陷。Claisen重排后产生的烯丙基苯酚或烯丙基苯甲醛等化合物往往能够发生串联反应[8-9],基于此,选择恰当的反应物,通过串联Claisen重排反应,有望成为构建异色满酮结构的一种新方法。
传统上的Claisen重排一般需要在超过200 ℃的高温下驱动,反应耗能大,且对反应底物的热稳定性提出了很高要求。通过添加Lewis酸作为催化剂能降低反应温度,但会带来分离的困难和后处理的环境问题[10-11]。相对于上述传统方法,使用封闭的水热体系来驱动Claisen重排具有操作简便、无催化剂添加、产物纯度更高和环境友好等优势。

Scheme 1
通过不同温度的水热反应条件筛选,最佳的5-烯丙氧基间苯二甲酸二甲酯转化为3-甲基-5-羟基-7-羧基异色满-1-酮(K1)的反应条件得到了建立和优化,在水热120 ℃及以上时,原料会发生Claisen重排并高纯度地转化为异色满酮分子。
1 实验部分
1.1 仪器与试剂
SGW X4型显微熔点仪;Anasazi EFT-60型核磁共振仪(DMSO-d6为溶剂,TMS为内标);Nicolet IS50型傅立叶变换红外光谱仪;Perkin-Elemental 2400型元素分析仪。
5-羟基间苯二甲酸二甲酯,九鼎化学(上海)科技有限公司;烯丙基溴,国药集团化学试剂有限公司,其余所用试剂均为分析纯或化学纯。
1.2 合成
将5-羟基间苯二甲酸二甲酯(1)1.02 g(5.7 mmol)、烯丙基溴0.68 g(5.7 mmol)、碳酸钾2.05 g(15 mmol)、碘化钾0.33 g(1.9 mmol)加入三口烧瓶中,加入丙酮30 mL作为溶剂。缓慢搅拌,升温至回流,反应4 h(TLC检测)。旋转蒸发除去丙酮,加入过量水洗涤后用二氯甲烷萃取并分液,合并有机层。旋转蒸发除去二氯甲烷后得到5-烯丙氧基间苯二甲酸二甲酯(2):白色固体1.09 g,产率87%;1H NMRδ:3.95(s,6H),4.63(d,2H),5.41(m,2H),6.05(m,1H) ,7.75(s,2H),8.25(s,1H);IRν:2960,1714,1646,1466,1603,768,867 cm-1。
取5-烯丙氧基间苯二甲酸二甲酯(2)0.2 g(0.9 mmol)装入水热反应釜中,溶解于10 mL去离子水中,160 ℃,反应72 h,而后以2.5 ℃/h的速率缓慢降温至室温,使转化后的产物慢慢以晶体形式析出,过滤,干燥后得白色晶体K10.183 g,产率91.9 %,m.p.134~136 ℃;1H NMRδ:1.49(s,3H),2.51~3.05(d,2H),4.59(q,1H),7.66(s,1H) ,7.98(s,1H),11.64(s,1H);IRν:3553,3183,2983,1682,1611,1593,1440,761,717 cm-1;Anal.calcd for C11H10O5:C 59.46,H 4.50;found C 59.86,H 4.41。
2 结果与讨论
2.1 合成条件与选择性
将5-羟基间苯二甲酸进行酯化并与烯丙基溴通过威廉姆森反应合成了5-烯丙氧基间苯二甲酸二甲酯。酯化反应的引入提高了在水热条件下底物的溶解性,相对于5-烯丙氧基间苯二甲酸为原料的反应,产物的纯度和底物的投料量都大幅度增加。随着Claisen重排反应的发生,烯丙基在迁移至邻位后会与邻位的羟基或者羧基发生进一步的亲核加成反应,分别生成内醚或者内酯,当羟基和羧基并存,双键的加成往往并不会表现出高的选择性,即便控制反应条件使其中一种产物呈现优势,也难以达到完全单一的转化[12-14]。
对于化合物2的反应而言,此时中间态3有发生两种加成的可能性,分别是与羧基上的氧原子形成六元环,最终稳定为3-甲基-5-羟基-7-羧基异色满-1-酮(K1)的结构,以及与酚羟基构成五元环,最终形成2-甲基-4,6-二羧基-2,3-二氢苯并呋喃(K2)的结构。通过对水热法所得到的产物进行纯度分析,其结构经1H NMR,FT-IR和元素分析确证,最终确认了在水热条件下5-烯丙氧基间苯二甲酸二甲酯会完全单一地转化为异色满酮的K1结构,从选择性上来看,这点与以往其他实验所报道的结果截然不同。同时,对所获得的产品的晶体进行单晶衍射分析,获得了K1的X-射线单晶衍射结构,进一步确认了化合物的分子结构和固态排列方式。
传统方法驱动类似的结构进行Claisen重排时,在不添加催化剂的前提下需要使用有机溶剂和200 ℃以上的高温,产率也仅有80 %左右[11]。对水热法而言,则只需较低的温度就能达成较高的转化。具体的转化温度对机理的探索以及反应活化能的计算具有非常重要的价值。为了探索能催化此反应的最低温度,我们以一个较低的反应物初始浓度,在160~110 ℃的区间每隔10 ℃设置一组对照试验,发现在110 ℃下反应釜的水相中总是存在尚未反应的原料,析出的晶体也存在明显的杂质。而水热温度在120 ℃及以上时,所得到的产物均为纯的K1晶体,水相中也未检测到原料的存在。
底物的浓度对转化的结果也有一定的影响,同时为了验证反应过程,我们用5-烯丙氧基间苯二甲酸(4)和2作为水热反应的原料进行对比,设置了一系列浓度并在120 ℃和160 ℃两个温度下进行实验,结果见图1。由图1可知,,反应物的浓度与温度共同影响着最终的转化结果,当浓度一定时,温度越高则越有利于转化的发生;当温度一定时,反应物的浓度也存在一个临界值,当浓度高于此临界值时,转化也不能彻底进行。同时我们发现当条件相同时,2、4两种不同原料所得的实验结果并无特别明显的区别。
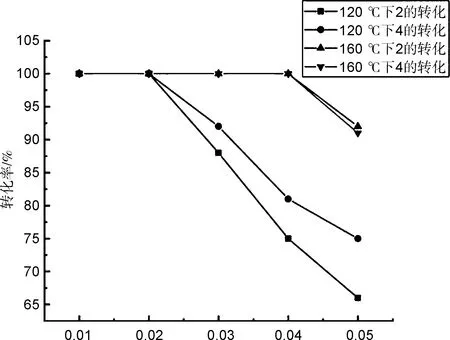
g/mL图1不同条件下2和4的转化率Figure 1Conversion rates of 2 and 4 under different conditions
K1作为异色满酮结构体系中的新成员,其结构中的羧基和酚羟基都属于活性基团,可以很容易进行取代或其他修饰,这使得K1作为药物合成的中间体或者原料具有很高的应用潜力。例如TAKAHASHI课题组[15]合成的诺伯根素衍生物和LEE[16]课题组合成的几类水解单宁,都含有K1的结构,显而易见都可以使用K1作为替代原料或者中间体来展开合成。
对本策略而言,水热条件下酯的水解-烯丙基重排-加成成环是一个串联反应,不需要额外的操作步骤,因此在不占据重排位点的前提下,也可以通过改变反应物的结构来直接控制最终得到产物的结构。
2.2 K1的晶体结构
选取合适尺寸的单晶,以φ-ω扫描方式收集衍射点。数据经经验吸收校正后使用Shelxtl软件包,采用直接法解出晶体结构。全部非氢原子坐标及各向异性热参数用全矩阵最小二乘法修正。非水分子上的氢原子的坐标由理论加氢方法得到。晶体的结构解析及精修通过SHELXL软件包完成[17],K1晶体CCDC为2027097。
如图2所示,固态下,K1结晶于单斜晶系P2(1)/c空间群,其最小不对称单元为一个K1分子,一个单胞中存在互为镜像关系的两个K1分子,这导致K1化合物的结晶为对映异构体而非单一手性的晶体,因此空间群并非手性空间群。基于共轭的影响,K1结构中的氧原子均处于统一平面中,异色满环上的手性碳原子(C9)及其所连接的甲基指向平面的外侧。在结晶时,K1分子A首先会沿着C3→C9方向反转180°,构成一个与原分子旋转对称的对应结构B,由于K1分子可以近似地看做是一个平面,A与B可以认为是两个互相平行的平面,这两个平行平面在纵向上交叉AB排列,而在横向上,则会偏转一定角度形成另一组平行平面,这两组平行平面之间的夹角为126.882°,如图3所示,这样重复排列下去,最终形成了K1的晶体结构。

表1 K1的晶体学数据Table1 Crystallographic data for K1
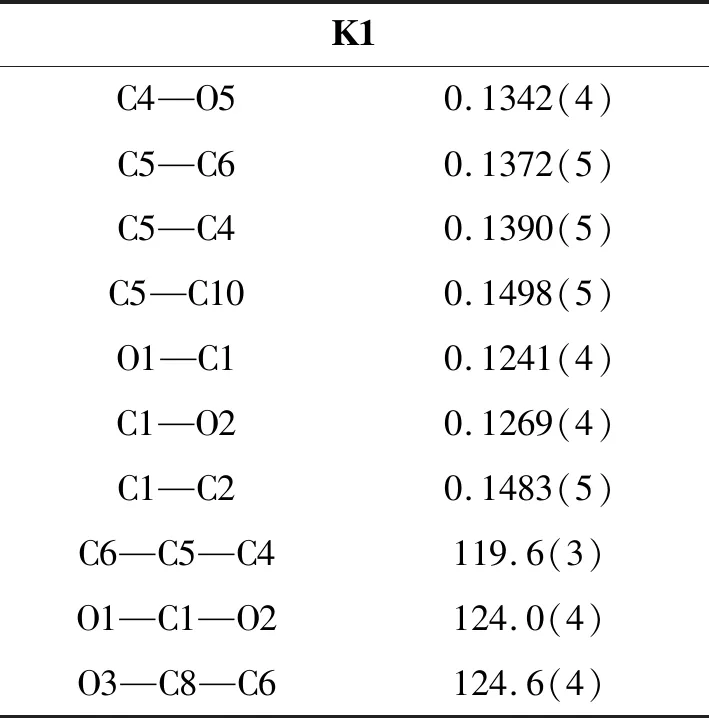
表2 K1的部分键长(nm)与键角(°)Table 2 Selected bond lengths(nm) and bond angles(°) for K1
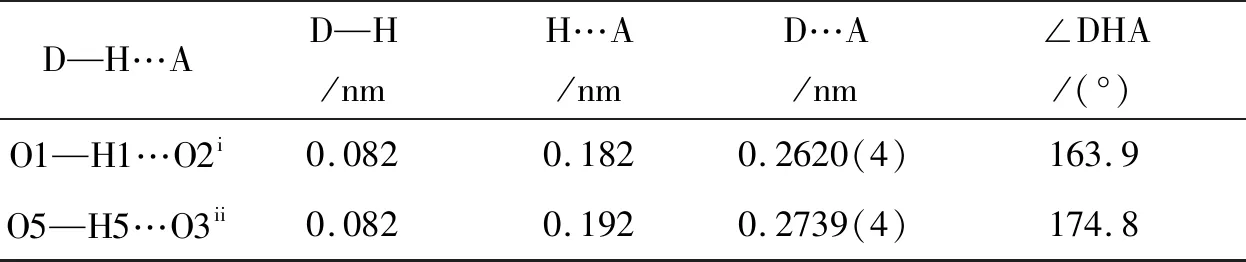
表3 K1的氢键键长(nm)和键角(°)Table 3 Hydrogen bond lengths(nm) and bond angles(°) for K1

图2固态下K1的单晶结构中的最小不对称单元Figure 2Minimum asymmetric structure of single crystal diffraction and enantiomeric parallel arrangement in K1

图3K1晶体的排列方式Figure 3Arrangement of K1 crystals
通过水热条件下的Claisen重排-亲核加成串联反应合成了含有异色满酮结构的化合物K1,研究了其化学结构和晶体结构,探究了在水热条件下可以发生完全转化的最低温度(120 ℃)和浓度(0.02 g/mL)。
猜你喜欢
橡塑技术与装备(2022年8期)2022-12-17
中国农业科学(2022年17期)2022-09-19
能源化工(2021年3期)2021-12-31
巴楚医学(2021年3期)2021-10-19
染整技术(2021年5期)2021-06-07
石油化工自动化(2020年1期)2020-03-05
中国循证儿科杂志(2019年2期)2019-06-04
安徽化工(2018年3期)2018-07-04
电脑知识与技术·经验技巧(2018年3期)2018-06-06
火控雷达技术(2016年1期)2016-02-06
