
结肠腺瘤患者与健康人肠液菌群的结构差异性分析
2021-03-10 06:20张蓓陈文晖汪妍
现代消化及介入诊疗 2021年1期
张蓓,陈文晖,汪妍
结肠腺瘤(colorectal adenomatous polyps, CAP)是起源于结直肠黏膜腺上皮的良性肿瘤,是常见的肠道良性肿瘤,被认为是最主要的癌前病变[1]。不同地区、不同年龄的发病率差别很大,40岁以下的发病率低,60岁以上较高,男女无明显差别。病因及发病机制尚不明确。肠道菌群是一个多样化的微生态系统,它是由超过2 000种细菌种类组成,并且拥有比人类基因组多出150倍的特殊基因组[2-3]。肠道微生物群可经内分泌、免疫、神经等途径来对宿主的消化吸收、能量代谢及免疫防御等基本功能产生影响[4,-5]。实践示,肠道菌群的稳态失调,对脂肪肝患者[6]、糖尿病患者[7]、炎症性肠病[8-9]患者[8、9]等患者等都有重要影响;现有研究认为肠道菌群和结肠腺瘤、结肠癌的发病也密切相关[10-12],但肠道菌群在结肠腺瘤、结肠癌中具体作用机制仍未明确,本实验研究从菌群的动态变化展开分析,探讨肠道菌群在结直肠腺瘤发生发展中的适应性改变,从而深入了解肠道菌群在结直肠腺瘤发病机制中的作用。
1 材料与方法
1.1 一般材料
本研究经过上海电力医院医学伦理委员会批准,选取2019年1月至9月上海电力医院消化内科结肠腺瘤患者10例(腺瘤组)和肠镜检查无异常的健康人12例(对照组),取其肠液样本。入组标准为:①年龄30~60周岁;②无肥胖以及超重的情况;③无胃肠手术史;④无遗传性非息肉性大肠癌及家族性腺瘤性息肉病;⑤无感染性疾病、慢性病、代谢性疾病;⑥肠镜检查前3个月内未用过抗生素、激素、益生菌;⑦无特殊饮食习惯;⑧纳入对象知情同意。选取单发的结肠腺瘤患者,部位分布:升结肠1例、横结肠2例、降结肠2例、乙状结肠3例、直肠2例,腺瘤直径在3~0.8 cm,结肠腺瘤患者均经组织病理明确为腺瘤性息肉。健康组取标本部位:升结肠2例、横结肠2例、降结肠2例、乙状结肠3例、直肠3例,均肠镜检查提示未见异常。标本采集:检查当日完善肠道清洁准备,晨5~6时口服复方聚乙二醇电解质2袋(冲泡2 000 mL液体),确定肠道准备完成后行无痛结肠镜检查,吸取肠液放入灭菌消毒后的EP管中,-40 ℃冰箱冷藏待检。
1.2 方法
对获取的肠液细菌16SrRNA基因序列V3-V4高变区片段进行精准的PCR扩增,PCR扩增应用的为NEB公司生产的Q5高保真DNA聚合酶,并对扩增循环数严格控制,使循环数尽可能低的同时,也确保同批样本扩增条件一致,扩增后对PCR产物定量检测,完成纯化后的混合DNA产物在lluminaMiSeq平台上完成高通量测序分析。为对原始双端测序数据进行整合,首先应用滑动窗口法,依次对FASTQ格式所对应的双端序列展开质量方面的筛查:窗口大小为10 bp,步长为1 bp,从5′第一个碱基处完成移动操作,要求窗口中碱基平均质量需≥Q20,自首个平均质量低于Q20的窗口处截断序列,要求截断后的序列长度需≥150 bp,另外,不允许存在模糊碱基N的情况。之后,利用FLASH软件,对通过质量初筛的双端序列根据重叠碱基进行配对连接:要求Read 1和Read 2序列重叠碱基长度需≥10 bp,并且碱基错配数小于重叠碱基长度10%。最后,依据单个样本所对应Index信息,将连接后的序列识别向对应样本分配(要求Index序列完全匹配),进而获得每个样本的有效序列(数据由派森诺生物有限公司提供)。
1.3 实验流程
微生物组总DNA提取→目标片段PCR扩增→扩增产物进行回收纯化→扩增产物实施荧光定量操作→测序文库制备→上机进行高通量测序。
1.4 统计学方法
将OTU的代表序列与Greengenes数据库(Release 13.8)相比对,使用QIIME软件,调用UCLUST这一序列比对工具(Edgar, 2010),按97%的序列相似度进行归并和OTU划分。然后,根据每个OTU在每个样本中所包含的序列数,构建OTU在各样本中丰度的矩阵文件(即OTU table)。计算物种累积曲线(Species accumulation curves)判断样本量是否足够并估计群落丰富度(Chao and Shen, 2004),当曲线趋于平缓时表明样本量已足以反映群落的丰富度,反之则表明样本量不足。使用丰度等级曲线(Rank abundance curve)了解群落物种的丰富度及均匀度,来反映样本的多样性。使用R语言工具绘制Specaccum物种累积曲线及丰度等级曲线。使用Metastats的统计学算法,在门和属水平对两组样本组间差异进行两两比较检验,找到两组之间的差异物种,使用错误发现率(False discovery rate,FDR)对P值进行校正,P值<0.05差异有统计学意义。将线性判别分析与非参数的Kruskal-Wallis以及Wilcoxon秩和检验相结合进行LEfSe(Linear discriminant analysis(LDA) coupled with effect size)分析,通过Galaxy在线分析平台(http://huttenhower.sph.harvard.edu/galaxy/),提交属水平的相对丰度矩阵进行LEfSe分析。根据LDA差异分析对数得分值(>2)和P(<0.05)值描述在组间具有显著差异的分类单元的统计学效力强弱。
2 结果
2.1 样本物种丰富度分析
Specaccum物种累积曲线(图1)示,物种累积曲线自样本量超过10以后逐渐趋于平缓,样本量达到15以后曲线已基本趋平,提示本次实验样本量已足以反映群落的丰富度。

图1 Specaccum物种累积曲线图 横坐标为实验样本量,纵坐标为被检测物种数,蓝色阴影为曲线置信区间
样本丰度等级曲线(图2)从横坐标看各样本绘制成的曲线在横轴上的跨度在300~1 000,跨度大,提示样本的群落的丰富度高,纵坐标观察曲线的下降趋势,图中各样本代表曲线在OTU值(经Log2对数转换)0~10表现为平缓下降,提示群落物种组成的均匀度较高。提示此次实验中样本群落多样性高。
2.2 两组肠液菌群门水平比较
所有样本在优势菌门方面均包括拟杆菌门(Bacteroidetes)、厚壁菌门(Firmicutes)和变形菌门(Proteobacteria),见图3。对照组丰度最高的前3个门依次是:厚壁菌门(46.5%)、拟杆菌门(32.38%)、变形菌门(17.31%);腺瘤组丰度最高的前3个门依次为:厚壁菌门(36.29%)、变形菌门(33.98%)、拟杆菌门(26.43%),厚壁菌门/拟杆菌门比值呈下降征象。Metastat分析显示,两组间厚壁菌门、变形菌门丰度比较,差异均有统计学意义(P<0.05)。
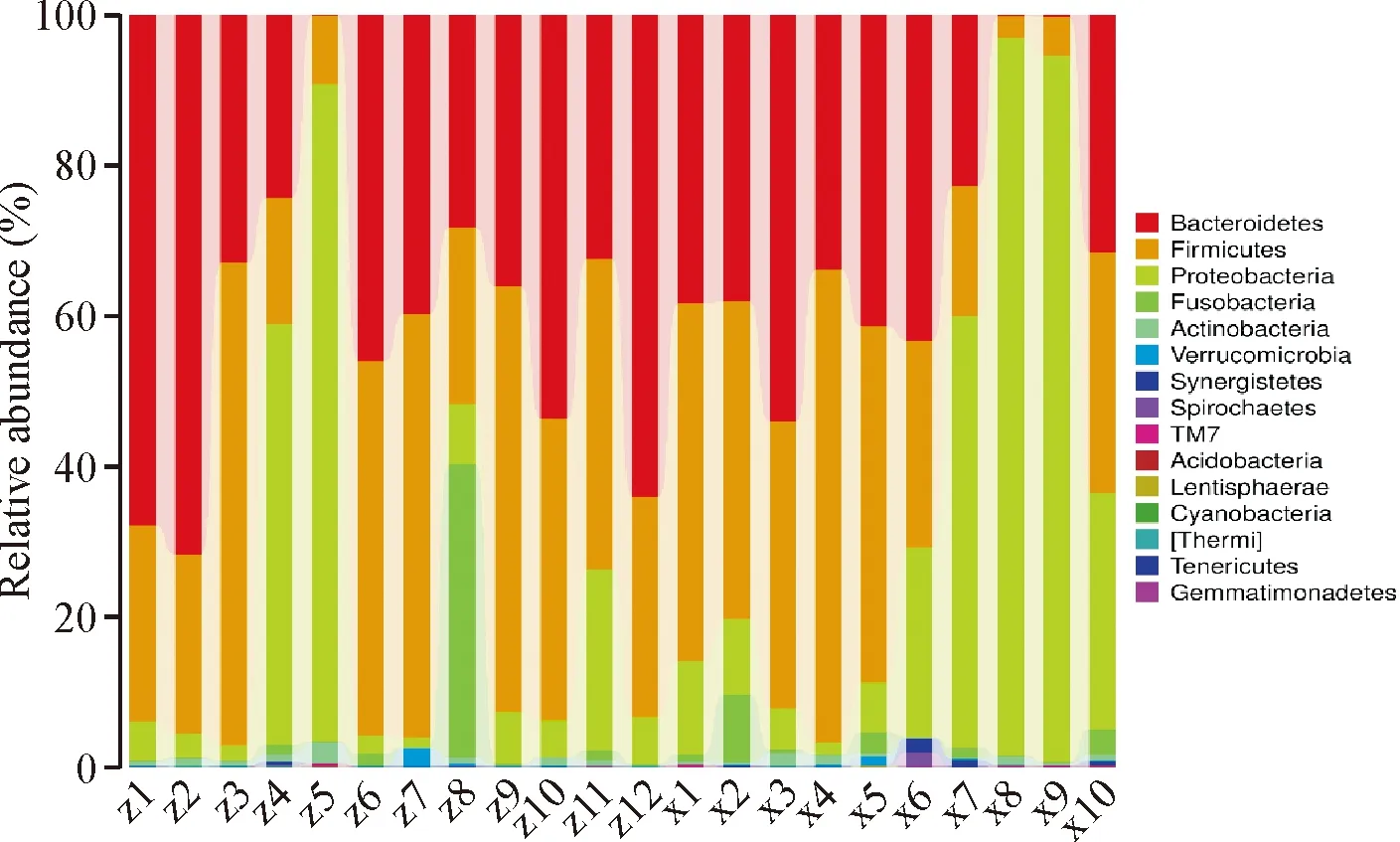
图3 两组样本在肠液菌群门水平上的相对丰度分布图(X腺瘤组,Z对照组)
2.3 两组样本肠液菌群在属水平上分析比较
两组样本肠液菌群属水平相对丰度见图4。Metastat分析显示,两组肠液菌群属水平上有13组菌序列量(绝对丰度)存在显著差异,丰度最高的菌属分别有假单胞菌属、拟杆菌属、杆菌属、肠球菌属、螺杆菌属等(图5)。LEfSe统计检验结果(表1),提示对照组中两组样本之间肠液菌群在属水平上假单胞菌属、杆菌属、肠球菌属存在显著差异(P<0.05)。其中对照组的肠液菌群中Erysipelotrichaceae在两组间有显著差异(P=0.048),属于肠球菌属,腺瘤组肠液菌群中Pyramidobacte、Dethiosulfovibrionaceae(P=0.007)、Pseudomonadaceae(P=0.013)、Gardnerella、Comamonas(P=0.048)等在两组间有显著差异,在细菌菌属上属于杆菌属、假单胞菌属。通过LDA值分布柱状图(图6),展示了LDA score大于预设值(2)、不同组中在丰度上有显著差异的物种,长度越长表明该有显著差异的物种影响越大,其中comamonas(假单胞菌)LDA差异分析对数得分值为4.286,为腺瘤组中有着显著差异的物种中影响最大的,而对照组中Erysipelotrichaceae是对照组中影响最大的显著差异的物种。
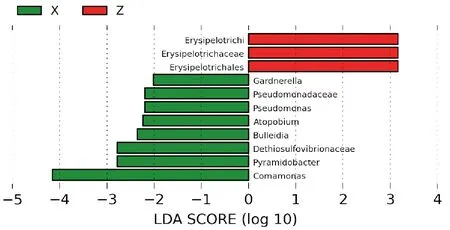
图6 LDA值分布图 绿色X腺瘤组,红色Z对照组;纵坐标为组间差异显著的分类单元,横坐标为LDA差异分析对数得分值
表1 LEfSe统计检验结果(仅组间差异显著者)
注:对数转换值为各分类单元具有的最高组内相对丰度均值,组别为具有统计学差异的分类单元

图5 两组肠液菌群属水平序列量比较 横坐标为差异显著的分类单元,纵坐标为各分类单元序列量。采用小提琴图结合箱线图的形式来呈现,“小提琴”的“胖瘦”可反映样本数据分布的密度高低情况;箱线图边框代表上下四分位数间距,横线代表中位值,上下触须依次代表位于上下四分位外的1.5倍IQR范围,符号“?”代表超过范围的极端值
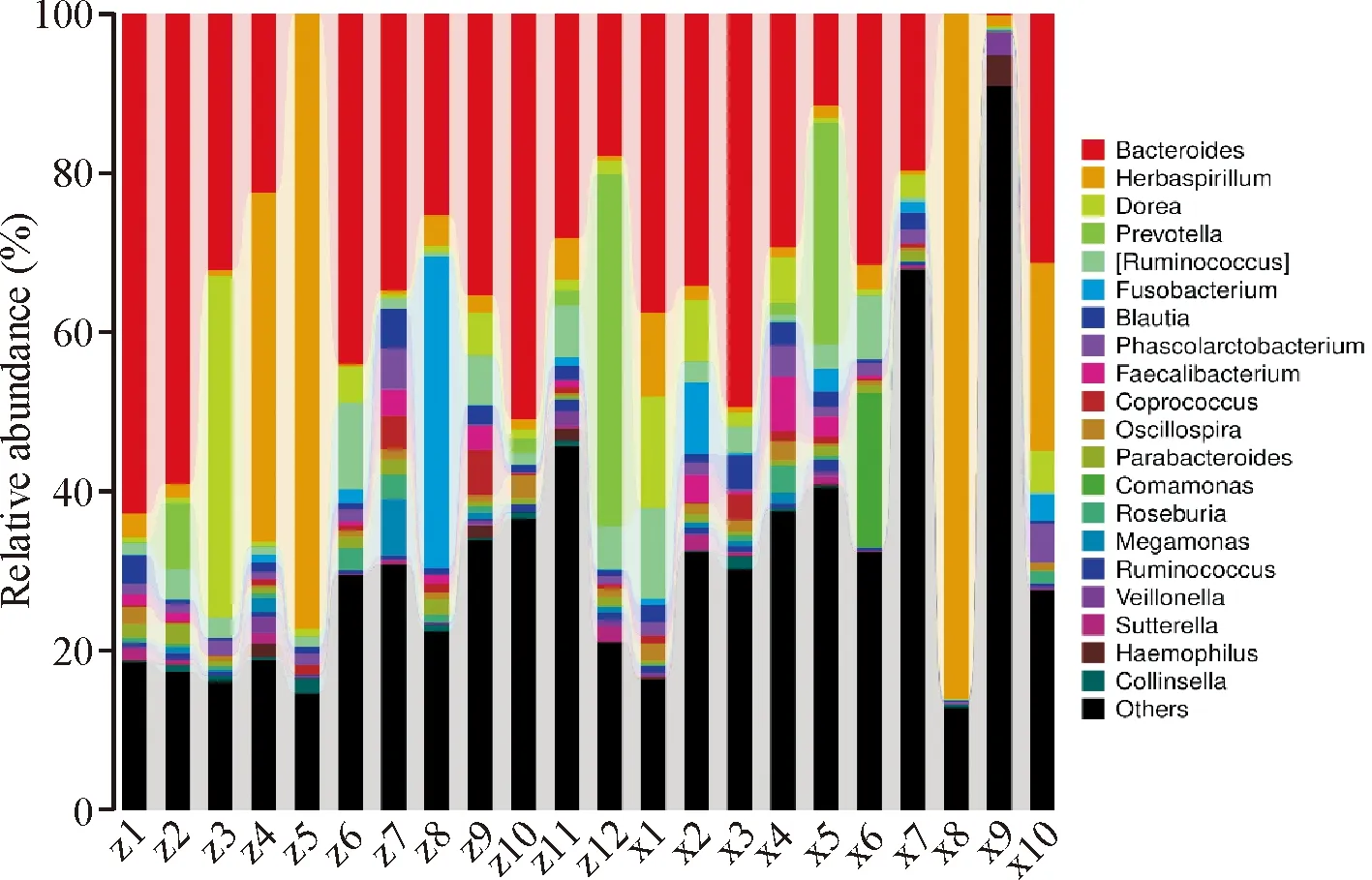
图4 两组样本在属水平相对丰度分布图
2.4 菌群比较PLS-DA分析
PLS-DA判别图(图7)提示同一分组样本点距离靠近,不同分组样本点距离远离,提示分类模型性能优良。
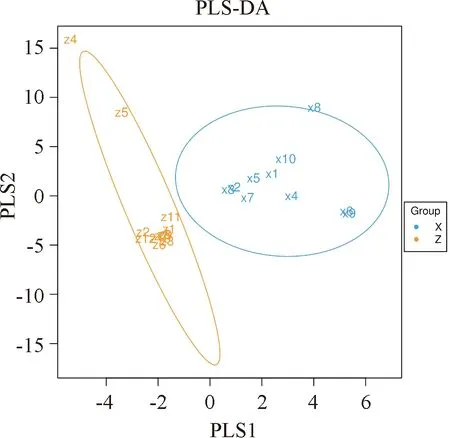
图7 PLS-DA判别分析图 单个点对一个样本,以椭圆标出相同分组的点;Z:对照组,X:腺瘤组
3 讨论
本研究显示,无论是对照组还是腺瘤组的肠液菌群检测分析结果中,厚壁菌门(Firmicutes)均表现为占主体地位。所有样本的优势菌门均包括厚壁菌门(Firmicutes)、拟杆菌门(Bacteroidetes)和变形菌门(Proteobacteria)。腺瘤组变形菌门的丰度显著升高(P<0.05),腺瘤组优势菌属为假单胞菌属、杆菌属,对照组优势菌属为肠球菌属。两组样本间肠液菌群结构组成在门、属水平上存在明显不同。肠道菌群定植肠道,构成肠道微生态平衡,对机体健康有重要的维护作用[13]。Baxter等[14]收集正常人及结肠肿瘤患者粪菌作研究材料,向无菌小鼠进行移植,并诱导肠癌模型,实验结果表明,肿瘤的数目与基线菌群间关联密切,即菌群的结构在一定程度上可决定肿瘤形成的易感性。Shen等[15]报道指出,临床腺瘤性息肉患者,肠道菌群中拟杆菌比例下降,而变形菌比例经观察呈升高状态。Goedert等[13]报道指出,腺瘤患者与健康人比较,在粪便菌群方面差异明显。
就门的水平展开分析,腺瘤组厚壁菌门较对照组占比明显下降,同时厚壁菌/拟杆菌门比值也表现为下降,而变形菌门占比则升高。变形菌门属于肠道菌群中较大的菌门,其中
γ-变形菌纲存在许多重要的已知病原菌及条件致病菌,其中部分条件致病菌如假单胞菌、部分杆菌,可释放对人体有害的内毒素[16],在腺瘤患者样本中表现为明显增加的情况。
腺瘤组中升高的假单胞菌属,其中部分菌种对人和动物存在致病性,相关研究表明,结肠癌患者在手术完成后,肠道中分布的假单胞菌含量明显增加[17-18],表明假单胞菌属与结肠癌发病可能存在相关性。虽然结肠腺瘤及结肠肿瘤发病机制是多方面的,并非单由菌群活动诱发,但菌群稳态失调可能在结肠腺瘤、结肠肿瘤病程中起到了重要作用,通过早期对肠道菌群实施高通量测序,根据肠镜菌群构成改变,可为早期筛检、诊治结肠癌前病变(结肠腺瘤)提供参考依据[19-20]。
综上,结肠腺瘤患者肠液菌群结构呈明显改变,形成了易于腺瘤生长的肠道微生态环境。通过优化菌群结构可能可以干预结肠腺瘤及其他结肠癌前病变的发生,对预防结肠癌发生发展有重要的临床意义。
猜你喜欢
科学技术与工程(2022年26期)2022-11-01
福建医科大学学报(2022年3期)2022-09-10
中国农学通报(2022年14期)2022-06-01
野生动物学报(2022年2期)2022-05-16
中国典型病例大全(2022年9期)2022-04-19
皮肤病与性病(2021年3期)2021-07-30
健康之家(2020年12期)2020-12-28
湖北农业科学(2019年22期)2019-12-23
健康博览(2017年10期)2017-12-05
医食参考(2017年12期)2017-04-01
