
优化阿魏酸印迹微球的制备条件及其分子识别性能
2021-03-08 02:56:26张夏红李锦辉
材料科学与工程学报 2021年1期
董 雁,张夏红,李锦辉
(1.龙岩学院化学与材料学院,福建 龙岩 364012; 2.固体废弃物资源化利用福建省高校工程研究中心,福建 龙岩 364012)
1 引 言
阿魏酸(ferulic acid,FA),是肉桂酸的一种衍生物,化学名为4-羟基3-甲氧基肉桂酸,是植物中的天然活性物质。人们最早在植物的种子以及叶子中发现阿魏酸。阿魏酸在中草药、蔬菜和水果中含量很大,尤其是在活血化瘀中药植物如升麻、川芎、当归、阿魏等中含量较高。阿魏酸具有抗氧化、抑菌消炎、抗血栓、抗癌、抗辐射和提高膜稳定性等药理作用[1],因此广泛应用在医药、农药、保健品及化妆品中。阿魏酸在植物当中的含量一般在0.03%左右。目前一般采用紫外分光光度法、毛细管电泳法、高效液相色谱法等办法对其进行测量[2-4]。过去多采用活性炭、大孔树脂、阴离子交换树脂、生物质等固体吸附剂对阿魏酸粗提取液进行分离[5]。这些方法存在选择性差,分离效果差,成本高等缺点。
分子印迹技术(MIT)于20世纪30年代起源于免疫学,是近些年来兴起的一种交叉学科技术[6]。它利用交联剂、功能单体和模板分子,在特定的条件下聚合得到产物。这种聚合物通常具有与模板分子相应的空间结构,形成的吸附位点能够与模板分子相匹配,形成在三维尺度上和模板分子相契合并且对其具有良好选择、“记忆”的空穴。“记忆”空穴与模板分子无论是在空间结构、作用位点及大小上都高度“契合”,具有特异识别功能,对模板分子能够快速、高效、特异性地选择吸附。MIT具有对模板分子优异的识别能力以及更强的稳定性,促使其在临床药物检测与分析、分离技术、仿生传感器等诸多方面具有十分良好的应用前景[7-14]。
本研究采用沉淀聚合法,以阿魏酸为模板分子,偶氮类的偶氮二异丁腈为引发剂,丙烯酰胺为功能单体,乙烯-丙烯酸共聚物的乙二醇二甲基丙烯酸酯为交联剂,制备对模板分子具有特异识别功能的阿魏酸印迹聚合物微球(MIPs)。研究模板分子、功能单体与交联剂以及引发剂等反应物之间的最佳反应比例、反应时间,及其选择吸附性。该方法操作简单,生成的MIPs球粒径均匀,有望为阿魏酸的分离和提纯提供新途径。
2 实 验
2.1 试剂与仪器
氮气,阿魏酸(FA,AR),乙二醇二甲基丙烯酸酯(EDMA),肉桂酸(AR),偶氮二异丁腈(AIBN,CP),乙腈(AR),甲醇(AR),乙酸(AR)和丙烯酰胺(AM,CP)。
STA449F3同步热分析仪,S-3400N扫描电子显微镜(SEM),UNIC-2000紫外可见分光光度计(UV-Vis)和IS10 傅立叶红外光谱仪(FTIR)。
2.2 实验方法
2.2.1制备MIPs 准确称取0.25 mmol FA作为模板分子放入锥形瓶中,并加入60 mL的乙腈以及1 mmol功能单体AM,在(20±5)℃下超声振荡1 h,使反应物充分作用。将锥形瓶取出放置在4 ℃冰箱中,静置,12 h过夜。之后取出锥形瓶,在碘量瓶中依次加入6.25 mmol的交联剂EDMA、36.6 mg的引发剂AIBN,在(20±5)℃下超声振荡5 min,后充N210 min,密封。然后,将锥形瓶置于恒温水浴锅中在60 ℃的温度下反应24 h。反应完成后,取出沉淀物(即未洗脱的MIPs)进行抽滤,将沉淀物用100 mL甲醇/乙酸混合溶液(V甲醇/V乙酸=6∶4)于90 ℃的恒温水浴中在索氏提取器中洗脱24 h,以将模板分子(FA)和未参与反应的物质脱除。洗脱后抽滤,并用甲醇洗去多余的乙酸,得到白色粉末状产物。烘干白色粉末状产物,得到的产物为MIPs,置干燥器中备用。
除不加入FA外,非印迹聚合物微球(NMIPs)制备的操作步骤与印迹聚合物相同。
2.2.2聚合物结构形貌及结构表征 采用SEM对微球的表面进行观察;用溴化钾压片法测定,在4000~400 cm-1范围内测定FA、MIPs、NMIPs及未洗脱的MIPs红外光谱,并进行分析。
2.2.3热分析法 用同步热分析仪在N2气氛下,对MIPs的热稳定性进行测试,升温速率控制在15 K/min,升温至800 ℃。
2.2.4MIPs的吸附性能研究 准确称取0.09 g的FA溶于乙腈中配置成浓度为0.9 g/L的FA标准乙腈溶液。分别称取第2.2.1节中制备的MIPs置于小烧杯中,准确量取15 mL FA标准乙腈溶液到装有MIPs的小烧杯中,在室温下超声震荡后,放置24 h离心分离。取1 mL离心后上层清液定容到25 mL的容量瓶。用乙腈为溶剂,在λ=320 nm测量其吸光度,按照图1及式(1)计算出相对吸附量Q(mg/g)[15]。
(1)
式中:C0为FA标准乙腈溶液的初始浓度,g/L;C为吸附后溶液中FA的浓度,g/L;V0为溶液的体积,L;m为MIPs的质量,g。
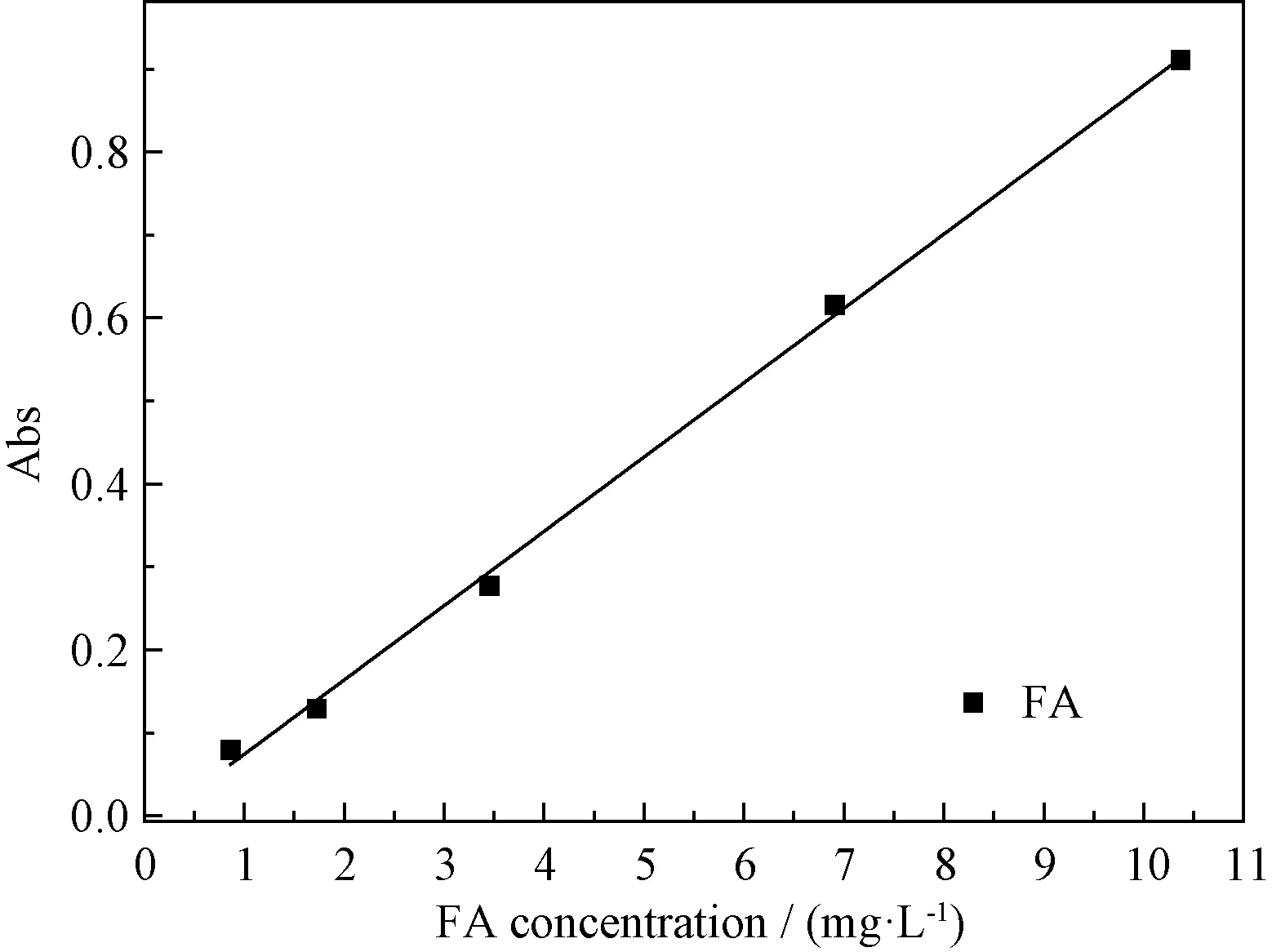
图1 阿魏酸标准溶液浓度(C)与吸光度(A)线性图Fig.1 Standard curve of ferulaic acid
2.2.5MIPs的选择吸附性能 选取结构与FA相似的肉桂酸为底物,采用静态平衡吸附法研究聚合物的选择吸附性[16]。称取2份MIPs置于由相同质量配制成的FA、肉桂酸标准乙腈溶液中超声震荡放置24 h,离心后取上清液1 mL于25 mL容量瓶中用乙腈定容。根据式(1)、(2)分别计算MIPs对FA和肉桂酸的特异性吸附量。
Q=QMIPs-QNIPs
(2)
FA和肉桂酸结构式见图式1。
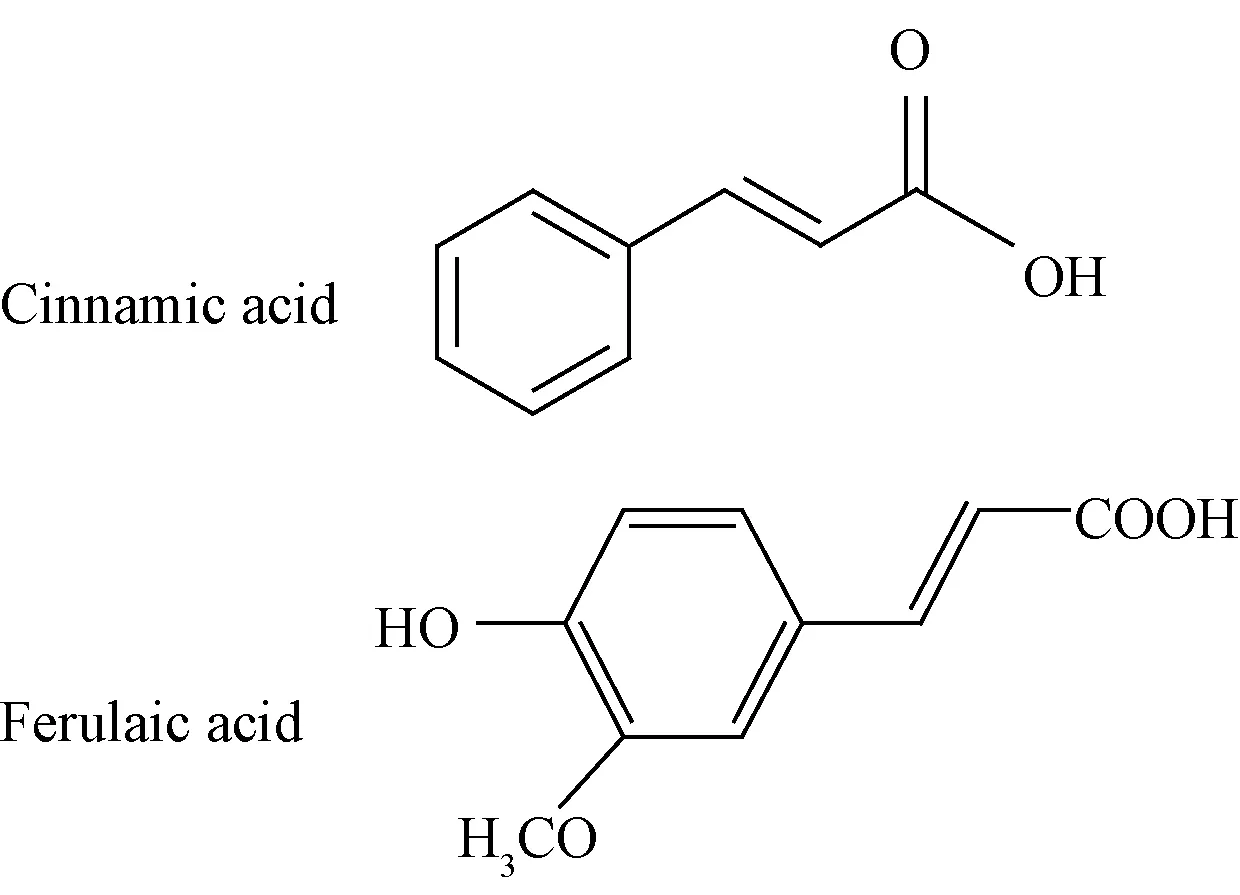
图式1 肉桂酸和阿魏酸的结构式Scheme 1 Structure of cinnamic acid and ferulaic acid
3 结果与讨论
3.1 MIPs和NMIPs的微观形貌观察
从图2可见,在相同的时间、温度及搅拌速率的条件下制得的MIPs与NMIPs,大小较均匀,球形度较好,外观无明显差异。MIPs和NMIPs在放大5000倍的电镜下形貌固然相似,但是MIPs形状更圆润,粒径分布均匀性较NMIPs好。其原因可能是FA会与AM形成氢键,使MIPs具有更多印迹“空穴”。这些“空穴”源于为FA“专属定制”的,因而空穴的三维网络空间大小及形状与FA完全契合,FA所含的特征基团与MIPs空穴内的表面基团高度互补,致使MIPs对模板分子具有强大的“记忆力”。

图2 聚合物的电镜扫描图片 (a)MIPs,(b)NMIPsFig.2 SEM images of (a) MIPs and (b) NMIPs
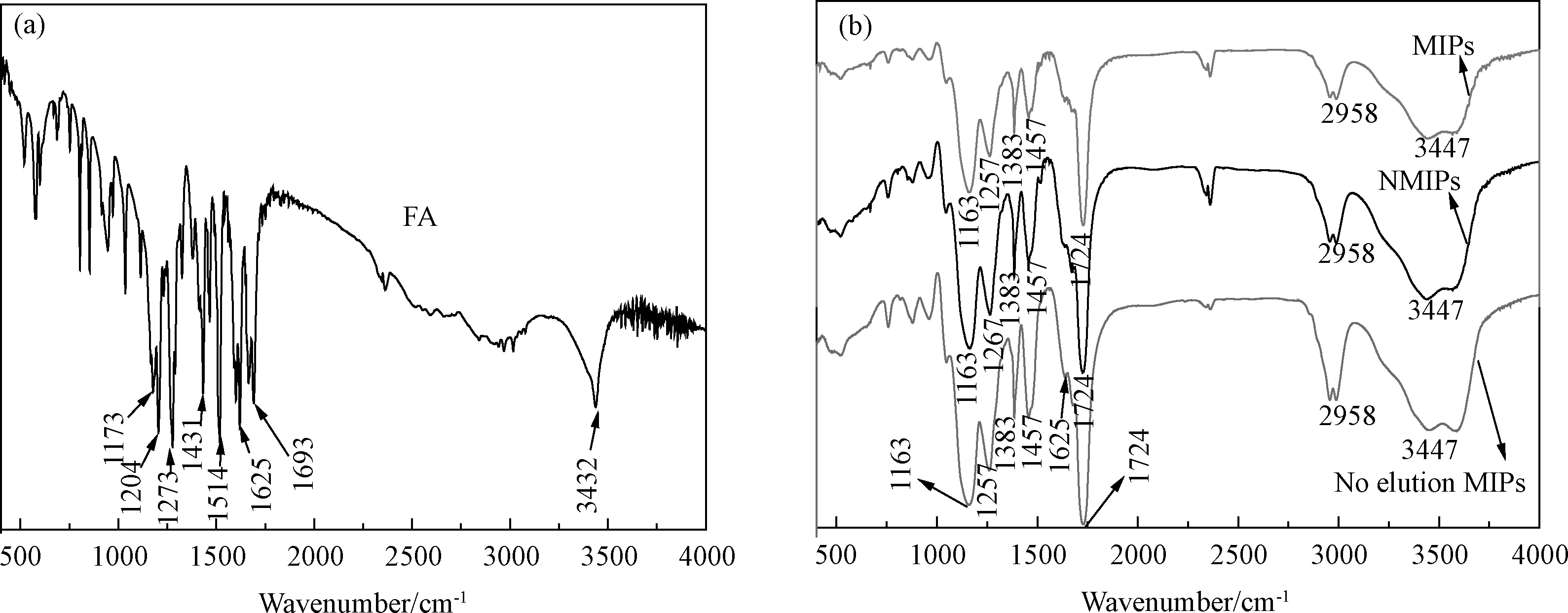
图3 (a)阿魏酸,(b)未洗脱MIPs,NMIPs和MIPs的红外光谱图Fig.3 FTIR spectrogram of (a)FA and (b) No elution MIPs, NMIPs and MIPs
3.2 红外光谱分析
图3给出FA、未洗脱MIPs、NMIPs及MIPs的红外光谱图。从图可见,FA与其他三种聚合物的红外光谱图存在显著区别。图3(a)中,FA的羟基的吸收峰在3432 cm-1处,羧基结构上-C=O的特征吸收在1693 cm-1处出现,在1625和1514 cm-1处出现的是苯环的吸收峰。从图3(b)上可见,AM骨架上的-C=O伸缩振动峰在1724 cm-1处,C-H伸缩振动峰在2958 cm-1处,伯胺基团上的N-H伸缩振动峰在3447 cm-1处出现。未洗脱MIPs中含有FA,FA的苯环骨架的振动峰在1625 cm-1,然而,NMIPs及MIPs在此处都无吸收峰,这说明NMIPs和MIPs中不存在模板分子FA,FA已经被完全除去。图3(b)中还可以知,MIPs的吸收峰相较于NMIPs的吸收峰更为尖锐,这意味着NMIPs表面可能有更多的游离-OH,这可能是由于氢键存在直接会影响羟基的振动,从而造成NMIPs的吸收谱较MIPs的宽。
3.3 热重分析
将MIPs和NMIPs完全干燥后的样品在STA-449 F3同步热分析仪中,N2氛围下进行热重实验,测量分析聚合物的稳定性。
从图4可见,在0~102 ℃,随着温度升高MIPs和NMIPs二者质量均呈下降趋势,这可能与温度上升,聚合物中存在的少量水分蒸发有关。在102~281 ℃,曲线呈现平稳的趋势,说明在该温度范围内微球质量保持相对稳定;在281~450 ℃,MIPs和NMIPs质量有明显的下降趋势,尤其在281~355 ℃,MIPs失重率比NMIPs失重率的下降速率大。造成这种现象的原因可能是MIPs表面存在大量的“记忆”空穴,在此温度条件下微球的破坏主要发生在表面,使得MIPs表面结构较NMIPs更疏松,即在此温度区间MIPs的热稳定性比NMIPs的低。在340~396 ℃,MIPs与NMIPs的质量下降速率基本相同。在396~450 ℃,NMIPs的质量下降速率大于MIPs的质量下降速率,这可能与在此温度区间微球的破坏发生在微球内部,从而减缓了高温对微球的破坏作用,使得微球质量减小的速率有所减缓有关。在450~800 ℃温度区间,随着温度升高,MIPs和NMIPs的失重率下降不多,表明微球基本完全燃烧,MIPs的失重率可达98.51%,NMIPs的失重率达98.32%。
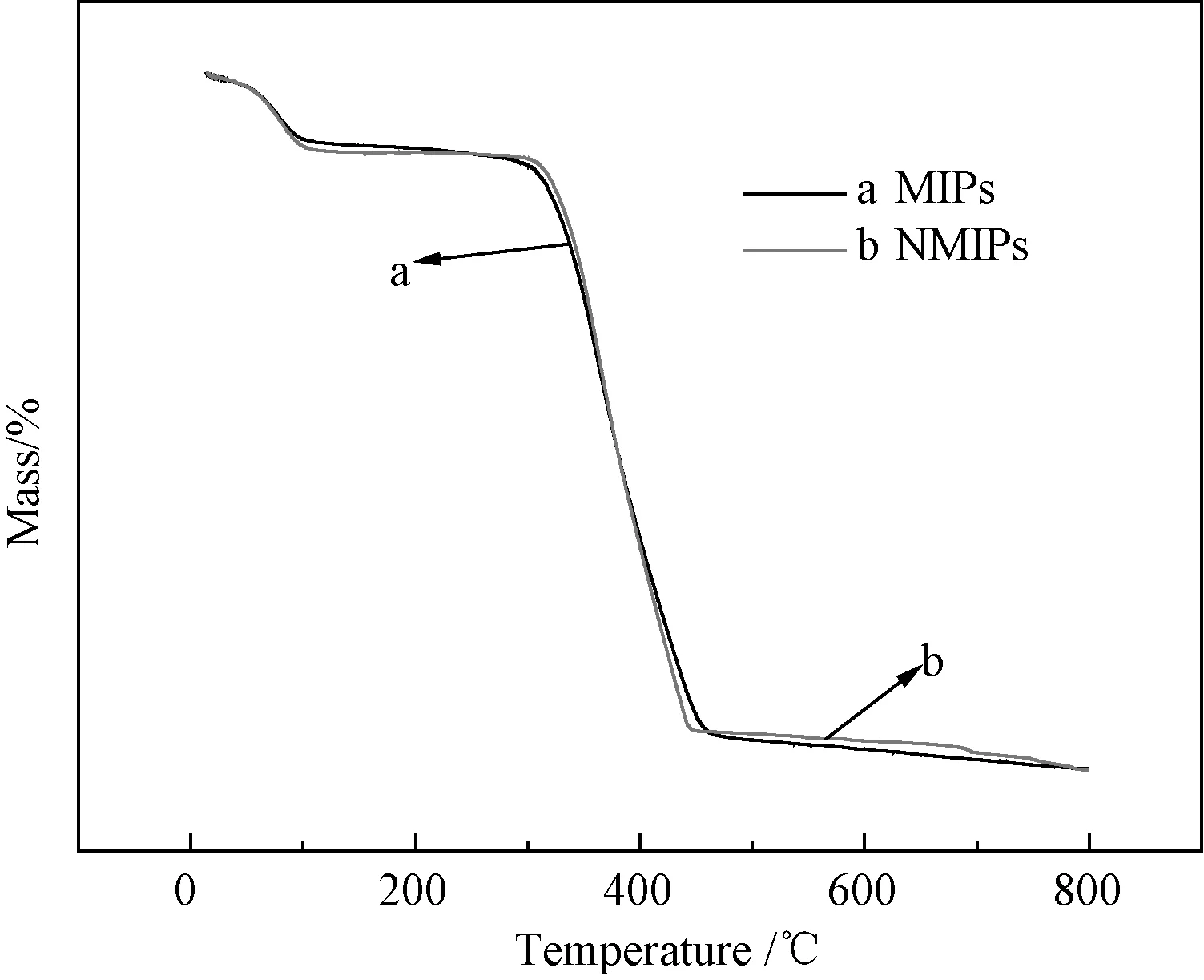
图4 聚合物的TG曲线Fig.4 TG curves of polymers
3.4 不同反应条件对MIPs吸附性能的影响
3.4.1AIBN用量对吸附性量的影响 从图5可以看出,随着AIBN用量增多MIPs的吸附量持续上升,当AIBN用量达到0.0366 g时MIPs的吸附量在该实验条件下达到最大值129.20 mg/g,之后随着引发剂用量增加其吸附量呈下降趋势。其原因是:在吸附量达到最大值之前,引发剂用量的不足使反应不完全,只有部分反应物参与反应,但随着引发剂用量的增加,生成的自由基逐渐增多,聚合速度加快,得到的MIPs增多,吸附量也增加;达0.0366 g到后,在功能单体用量固定的情况下,引发剂用量增加,反而会形成一些结构不完整的印迹位点,致使MIPs的吸附性能下降。
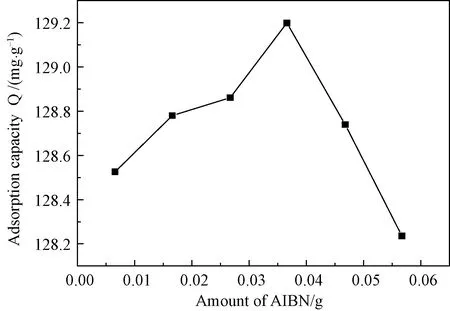
图5 AIBN对MIPs吸附性能的影响Fig.5 Influences of Different Amounts of AIBN on Adsorption property of MIPs
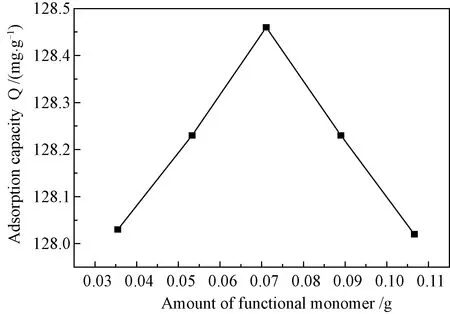
图6 功能单体AM的相对用量对MIPs的吸附性能的影响Fig.6 Influences of different amounts of functional monomer on adsorption property of MIPs
3.4.2功能单体AM用量对吸附性能的影响 由图6可以看出,随着功能单体AM用量的增加,MIPs的吸附量上升。当AM量超过0.07 g后,吸附量达到最大值为128.460 mg/g。之后,随着AM用量增加,吸附量反而降低。原因或许是在AM用量较少时,绝大多数的模板分子未能参与反应,少量的AM与FA形成络合物,起初所形成的MIPs量少,且MIPs中具契合模板分子“空穴”数目也很少,吸附量不高;随着AM用量的增加,MIPs中具有契合模板分子 “空穴”增多,对FA的吸附量显著增加,直到最佳值。但是,由于模板分子数量有限,要形成有效的“空穴”过量的功能单体反而会造成一定干扰,促使MIPs的识别能力变差,从而使吸附量降低[17]。
3.4.3EDMA量对吸附量的影响 从图7可见,MIPs吸附量随着EDMA量的增加先表现为上升的趋势,当交联剂用量达到1.24 g后,MIPs的吸附量达到最大值128.91 mg/g,之后随着交联剂用量增加吸附量而呈现下降趋势。开始时吸附量增加,可能与使用的交联剂较少,使得制备的MIPs不具有很好的交联度,微球的物理性能较差,以及在制备及洗脱过程中对印迹空穴造成破坏,使得微球中FA空穴的结构不完整,吸附性能低有关。随着交联剂的增加,其交联度逐渐提高,吸附量增加,达到吸附饱和状态后再加入交联剂,则会使微球交联度密度提高,导致微球结构过于紧密从而给洗脱带来困难,反而导致微球的吸附性能下降。
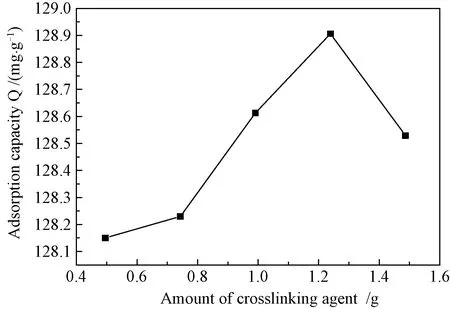
图7 交联剂的相对用量对MIPs吸附性能的影响Fig.7 Influences of different amounts of functional monomer on adsorption property of MIPs
3.4.4反应时间对吸附量的影响 从图8可见,MIPs的吸附量随着反应时间的增加而提高,当反应24 h时达最大值,之后基本稳定。其原因是MIPs和空穴数量也会随着反应时间增加,当反应时间达到24 h时,反应基本完成,空穴数量不再增加,吸附趋于稳定。
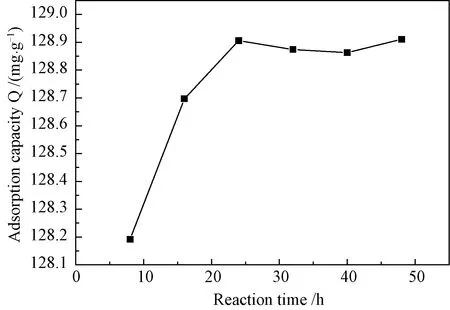
图8 不同反应时间对MIPs吸附性能的影响Fig.8 Influences of different reaction time on adsorption property of MIPs
3.5 MIPs选择吸附性能的研究
采用与FA结构类似的肉桂酸作为竞争底物,采用动态平衡吸附法,研究MIPs对模板分子FA的选择识别性能。按式(3)计算MIPs对底物的特异性吸附量:
Q=QMIPs-QNMIPs
(3)
式中:QMIPs和QNMIPs分别为MIPs与NMIPs对底物的最大吸收量,将 MIPs对Fa的选择因子α定义为1,其它底物(肉桂酸)的选择因子α定为MIPs对其它底物特异性吸附量与对FA的比值[18-19]。其中当α越接近于1则说明MIPs对底物的选择吸收性越好。
由表1可以看出,MIPs对FA的特异吸附量为175.467 mg/g,大于对其结构类似肉桂酸的吸附量(113.360 mg/g),这表明MIPs对模板分子FA具有良好的选择性,MIPs中的“空穴”与FA匹配,表现出很高的吸附性。由于肉桂酸与FA结构较为相似,因此MIPs对肉桂酸也表现出一定程度的吸附。但由于肉桂酸与FA结构略有不同,不能完全匹配FA的印迹“空穴”,因此MIPs对肉桂酸表现为较低的吸附性。由表1还可以看出,NMIPs对FA的吸附量为100.529 mg/g,对肉桂酸的吸附量为98.269 mg/g,非常接近,说明NMIPs对FA及其结构类似物不具有选择性。
3.6 MIPs等温静态吸附曲线
采用等温静态平衡吸附法研究吸附性能。在实验过程中配置浓度0.1~1.6 mg/mL一系列浓度梯度的阿魏酸乙腈标准溶液,将MIPs与NMIPs分别在这一系列标准溶液中吸附24 h后,测定溶液中阿魏酸浓度,得到静态吸附曲线,如图9所示。

图9 MIPs与NMIPs的静态吸附曲线Fig.9 Static adsorption curve of MIPs and NMIPs
从图可见,在0.1~1.6 mg/mL浓度范围内,随着FA溶液浓度的增大,MIPs与NMIPs的吸附量都得到提高,然而,在较高浓度下MIPs的吸附量显著高于NMIPs。这表示MIPs和NMIPs在空间结构上存在明显的差异。MIPs中加入了模板分子FA,因此存在与FA分子匹配的“记忆空穴”,而NMIPs中未加入模板分子,因此不具备与FA分子匹配的“记忆空穴”,MIPs吸附能力远大于大于NMIPs。
采用Scatchard模型研究MIPs对模板分子的结合特性。Scatchard方程[20]为:
(4)
其中,Qmax为结合位点的最大表观结合量(mg/g),Kd为结合位点的平衡离解常数(mg/mL),C为FA在上层清液中的平衡浓度(mg/mL)。
以Q/C对Q做图可以得到Scatchard分析图,见图10。
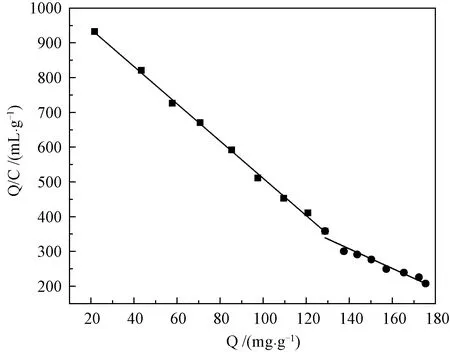
图10 MIPs的Scatchard分析曲线Fig.10 Scatchard Plots of MIPs
从图可见,Q/C对Q整体呈非线性关系,表明MIPs对FA的结合位点是不等价的。当Q分别为21.610~128.721 mg/g和128.721~175.467 mg/g时,Q/C与Q呈线性关系。以Q/C对Q作图,根据式(4)以可以求得高亲和位点的离解常数为Kd1=0.187 mg/mL,Qmax1=195.572 mg/g;低亲和位点的离解常数Kd2=0.354 mg/mL,Qmax2=248.724 mg/g。这显示0.1~1.6 mg/mL浓度范围内MIPs对FA存在不同的结合位点,具有不均一性[21],可能是由于模板分子FA结构中存在的-COOH和-OH能同时与AM发生络合,导致在交联反应以后形成了两种显著差异性的结合位点。如图11所示。
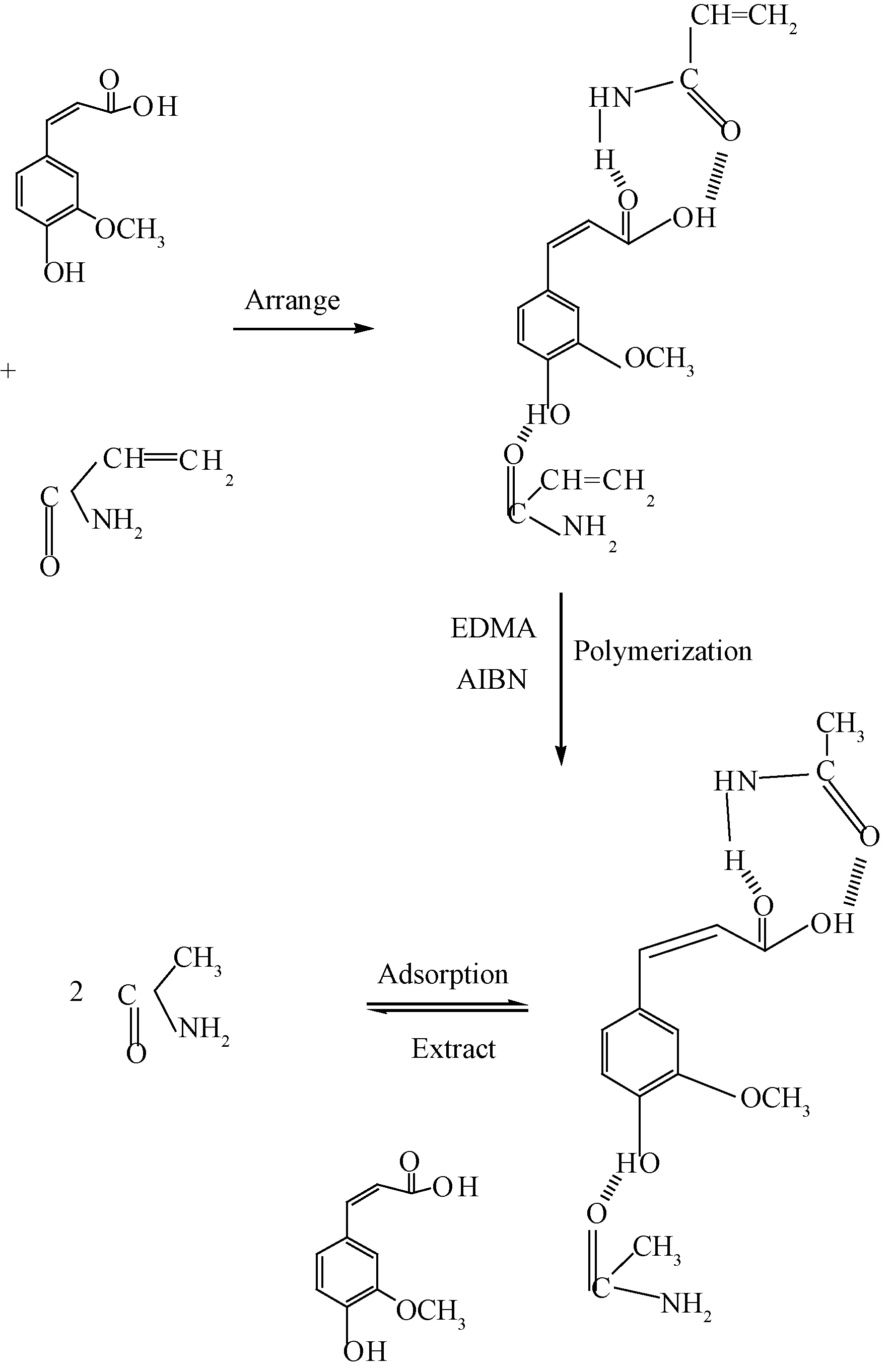
图11 阿魏酸印迹示意图Fig.11 Chemical process to imprint polymer with ferulaic acid
4 结 论
采用沉淀聚合法制备了MIPs,通过筛选反应条件,结构及性能表征,以及吸附性能的测试,得到如下结论:
1.制备MIPs最佳的反应条件是AM用量为0.07 g,EGMA为1.24 g,AIBN用量为0.0366 g,反应时间为24 h。所制备的MIPs粒径均一,球形度好。
2.MIPs对模板分子FA具有较高的选择吸附性,NMIPs对底物的识别不具备选择性。通过Scatchard分析得到高亲和位点的离解常数为Kd1=0.187 mg/mL,最大表观结合量Qmax1=195.572 mg/g;低亲和位点的离解常数Kd2=0.354 mg/mL,最大表观结合量Qmax2=248.724 mg/g。MIPs与NMIPs的热稳定性相差不大。
猜你喜欢
陶瓷研究(2022年3期)2022-08-19 07:15:18
云南画报(2021年10期)2021-11-24 01:06:56
粘接(2021年2期)2021-06-10 01:08:11
世界科学技术-中医药现代化(2021年10期)2021-03-02 05:51:44
小学生优秀作文(高年级)(2018年4期)2018-09-11 01:23:22
材料科学与工程学报(2016年4期)2017-01-15 13:35:48
石油化工(2015年9期)2015-08-15 00:43:05
橡塑技术与装备(2015年7期)2015-07-03 12:18:01
中国摄影(2014年12期)2015-01-27 13:57:04
华东理工大学学报(自然科学版)(2014年1期)2014-02-27 13:48:32
