
放射性污染土壤中铯的吸附/解吸行为研究进展
2021-03-06 05:27:22王善强李战国赵红杰韩梦薇
原子能科学技术 2021年3期
张 坤,王善强,李战国,齐 圣,赵红杰,韩梦薇
(1.国民核生化灾害防护国家重点实验室,北京 102205;2.国防工程研究院,河南 洛阳 471023)
核试验、核事故、核设施退役、核技术应用等人类核活动向环境中释放了大量的放射性核素,导致生态环境(空气、土壤、水体、植被等)受到不同程度污染[1]。放射性铯(Cs)在铀核裂变中产额大,是环境放射性污染的重要评价指标,也是人类活动释放到陆地生态环境中最重要的放射性核素[2]。近年来,放射性Cs在陆地生态系统中的吸附、解吸、迁移、转化、归趋等环境化学行为受到了广泛关注。
土壤和沉积物中的黏土矿物是土壤、沉积物、基岩等物质最重要的组成部分,是控制放射性Cs环境化学行为和归趋的重要固态介质,黏土矿物是控制放射性Cs在土壤中吸附、解吸、扩散、迁移等环境化学行为的关键因素。上述环境化学行为又进一步受到环境中不同介质之间的物理、化学、生物作用影响。放射性核素的地球化学性质受到土壤中的胶体颗粒、腐植酸、金属离子、微生物等多种因素影响。为全面了解和系统评估土壤中放射性Cs的环境化学行为,需对土壤中放射性Cs的吸附/解吸行为及其作用机制开展系统研究。本文拟在文献调研的基础上,对放射性Cs在黏土矿物中的吸附/解吸行为及其表征方法进行比较和总结,并就未来的发展进行展望,为放射性污染修复治理、核设施退役废物地质处理与处置等研究提供参考。
1 放射性Cs的来源
自1945年以来,全世界共开展了2 000多次核试验,核试验造成全球范围核污染扩散,对地球环境和生态系统造成长期危害[3]。土壤中放射性核素的性质和来源列于表1,放射性Cs主要有137Cs和134Cs两种同位素。切尔诺贝利和福岛核电站事故给生态环境造成极大危害[4],切尔诺贝利核事故造成4 300 km2范围内的土壤受到137Cs、90Sr、154Eu、238Pu、241Am等多种放射性核素不同程度的污染,事故导致大量放射性物质扩散并影响其他国家,如白俄罗斯、俄罗斯、德国、奥地利、希腊等国家[5];日本福岛核事故造成周边1 778 km2陆地范围内建筑物、道路、土地、森林植被污染,137Cs污染土壤总量达1 600万~2 200万m3[6]。2次事故后的监测结果表明,切尔诺贝利事故对环境的辐射后果影响远超过福岛事故,该核事故使全球区域的森林、土壤和河流、海域等受到不同程度的污染;日本福岛核事故释放的大多数放射性核素沉降于近海并沉积在太平洋。

表1 土壤中主要放射性核素性质与来源Table 1 Property and source of main radionuclide in soil
Cs是一种碱金属,与K的化学性质类似。核能利用导致大量的放射性Cs同位素释放于环境,特别是长寿命放射性核素137Cs(T1/2=30.2 a)[7]。137Cs通过β-衰变生成137Bam(T1/2=2.55 min),约占总β-衰变的95%,能量为0.51 MeV,随后衰变为稳定的137Ba,约占总β-衰变的5%,能量为1.17 keV。137Cs在铀核裂变中的产额大,生物利用度高,可通过生态系统和食物链危及人类健康。因此,137Cs污染已成为当今国际社会高度关注的问题。
2 黏土矿物结构与特性
常见的黏土矿物属层状构造硅酸盐矿物,由硅氧四面体和铝氧八面体构成[8-9]。黏土矿物可变负电荷层结构是其对阳离子吸附、解吸、生物有效性、扩散、传输的重要理论依据。永久负电荷或层间电荷通过同晶取代作用产生,主要来源有两种方式:一是四面体片(T)中的Si4+被Al3+取代;二是八面体片(M)中的三价阳离子(Al3+、Fe3+)被Mg2+取代。通过此类同晶取代作用,T-M-T层间被更多可交换阳离子占据,这种负电荷结构是2∶1型层状硅酸盐黏土矿物的重要特性之一[10]。
理想的1∶1高岭石(二八面体)中,Si4+被Al3+取代,则高岭石的分子式为(Si3Al1)-Al4O10(OH)8,由于高岭石外表面附近的碱性阳离子Na+和K+的平衡作用,高岭石的净电荷应为-1。但1∶1型层状硅酸盐的层电荷通常接近于0,这是由于层间阳离子交换作用所导致的。可变负电荷特性在2∶1型层状硅酸盐和云母类矿物中较为明显,2∶1型蒙脱石和云母的负电荷范围为0.2~1.0[11]。
此外,层间膨胀性是影响黏土矿物对放射性Cs吸附性能的另一重要因素,伊利石和绿泥石为非膨胀性矿物,蛭石为中等膨胀,蒙脱石具有较大的膨胀性,绿泥石层间位置被氢氧化物(Al(OH)3)所占据,蛭石和蒙脱石净电荷分别为0.6~0.9、0.2~0.6,伊利石净电荷最大(0.9~1.0),其层间的固定能力最强。黏土矿物的膨胀性有助于层间阳离子交换能力和放射性Cs的吸附亲和力提高。如通过考察伊利石、蛭石、蒙脱石和绿泥石对Cs的吸附能力表明,其对Cs的吸附能力大小顺序为:蛭石>伊利石>蒙脱石>绿泥石[12]。
3 放射性Cs在黏土矿物中的吸附/解吸行为与过程
3.1 吸附位点
黏土矿物吸附放射性Cs的位点类型与结合形态可分为三吸附位点和五吸附位点,如图1所示。三吸附位点包括:基面位点、楔形位点和层间位点。基面位点对Cs+的亲和力最弱,选择性差;风化作用形成的楔形位点对低水合能的Cs+具有较强的亲和性和较高的选择性(相对于其他碱金属和碱土金属离子);层间位点由楔形位点的Cs+扩散形成,对Cs+的亲和性和选择性仅次于楔形位点(图1a)[13-15]。五吸附位点包括基面位点、边缘位点、层间位点、楔形位点和水合层间位点(图1b)[16],边缘位点吸附特征与基面位点相似,水化夹层的吸附可促进楔形位点吸附,Cs+在黏土矿物中的吸附主要通过离子交换反应实现[17]。
3.2 吸附作用机制

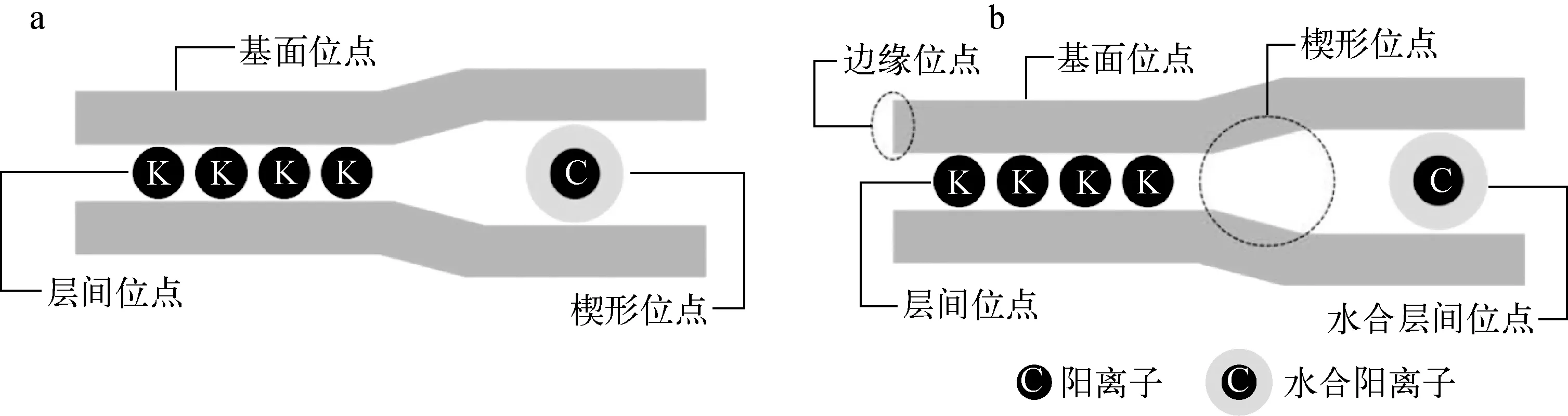
图1 黏土矿物中Cs+吸附位点示意图Fig.1 Schematic diagram of Cs+ adsorption site on clay mineral
Park等[20]提出了Cs+在伊利石上的吸附、迁移和非可逆性固定机制,如图2所示。一种非可逆固定机制是Cs+使楔形位点塌陷(图2a),Cs+选择性吸附在楔形位点上,Cs+吸附后楔形位点塌陷,楔形位点结构与正常夹层或边缘位点尺寸接近,Cs+迁移扩散至更深的层间区域;另一种作用机制是离子水合作用,由于K+的水合能高于Cs+,水合后的Cs+选择性吸附在楔形位点上,楔形位点上的水合Cs+脱水,楔形位点附近中间层的K+被水合,从而使Cs+和K+的位置发生置换,脱水后的Cs+在中间层中最终变为非可逆固定(图2b),这两种作用机制可充分解释Cs+在伊利石中与楔形位点和层间位点的选择性吸附与非可逆性过程。
3.3 测定黏土矿物中楔形位点容量的方法
通常采用放射性Cs截留电位(RIP)法和银硫脲(AgTU)法测定黏土矿物中楔形位点的容量。AgTU法是利用AgTu与黏土矿物基面位点结合并具有很强吸附亲和力的特点,假设所有基面位点因与AgTu的络合作用而被完全占据[18],Cs+只吸附在楔形位点上[21]。因此,利用过量AgTU可估算楔形位点吸附容量[22]。RIP法[23]与AgTU法原理相同,选用Ca2+和K+作为Cs离子取代阳离子。Wauters等[24]利用Ca2+和K+混合物进行了RIP测定,确定了楔形吸附位点含量。
AgTU法和RIP法主要通过分别添加高浓度的AgTU和Ca2+,使土壤黏土矿物的基面位点被AgTU和Ca2+吸附,而Cs+只能选择性吸附于楔形位点,楔形位点吸附容量约占土壤黏土矿物中总CEC的0.25%~1.76%。AgTU法是土壤中特定位点数量选择性吸附定量分析的技术基础,与AgTU法相比,RIP法操作简便且精确度高,更常用于估算黏土矿物中楔形位点的含量。
3.4 吸附模型
1) 等温吸附模型
等温吸附模型是基于平衡态和恒温态下液相和固相核素浓度之间的数学关系,常用吸附模型有Langmuir模型、Freundlich模型、Langmuir-Freundlich(L-F)模型和Dubini-Radushkevich(D-R)模型[25]。
Langmuir模型假设基于平衡状态下吸附和解吸分子数量在单位时间内相等,忽略矿物表面上的横向吸附相互作用和水平迁移的影响,表面吸附位置分布均匀;Freundlich模型是描述非均匀相吸附体系的经验模型,若固体表面不均匀,吸附平衡常数与表面特性有关;L-F模型是两个模型的结合。

图2 Cs+在伊利石上的选择性吸附和迁移过程 Fig.2 Selective adsorption and migration of Cs+ on Illite
上述4个模型已用于研究放射性核素在黏土固液界面的吸附与解吸行为。Shahwan等[26]的研究表明,Cs在高岭石、斜发沸石、膨润土上的吸附与Freundlich和D-R模型吻合较好。花岗岩对Sr的吸附符合Langmuir模型,而花岗岩对Cs的吸附为非线性[27]。批式吸附实验表明,红壤对Cs+的吸附与Freundlieh等温模型吻合较好[28]。
吸附等温模型在金属离子的吸附作用宏观研究中应用很广泛,实验中不考虑酸碱度对吸附的影响,也不能描述吸附相表面结构信息、金属离子在吸附相表面的化合态、表面吸附位点、累积浓度等适用范围。因此,经验吸附模型不能较好地解释放射性核素与黏土矿物界面吸附与解吸的作用机理。
2) 表面配位模型
表面配位模型(SCM)是一种考虑表面电荷作用、吸附剂表面位点和吸附质分子的特异性、表面配合物分子的形态、质量作用规律、物质平衡规律等多种微观和宏观作用机制的描述方法[29]。
Schindler等[30]提出了配位化合物分子的描述方法,证明了阳离子在表面带正电性吸附的可能性,表面配位模型既考虑了溶质在表面位点的内亲和力作用,又关注了带电表面与溶解离子之间的库仑相互作用。
两性表面水化羟基吸附是pH值影响的结果,而非土壤矿物溶解或H+交换[31],因此,表面配位作用机制可用于描述痕量Cs+在两性羟基上的吸附。如,Gutierrez等[32]利用三层模型(TLM)模拟了Cs+在Ca基蒙脱石上的吸附,层间和楔形边缘位点是Cs+吸附的主要原因,吸附的作用机理是边缘部位的特异性吸附,边缘吸附位点仅占总吸附位点的5%,但其吸附总量占比为94%。Silva等[33]提出了蒙脱石的另一种组合方法:(1) 具有1-pK双层模型(DLM)的双吸附位点(≡SOH和≡TOH基团);(2) 单位点阳离子交换模型,考虑了Cs+和Na+在≡SOH和≡TOH基团上的配位作用,该方法的缺点是需用12个参数进行拟合,应用过程复杂。Hurel等[34]利用一种表面配位模型研究了膨润土对阳离子(Cs+、Na+、K+、Ca2+和Mg2+)的吸附,利用硅醇边缘位点和阳离子交换模型模拟了Cs+的吸附过程。Wang等[35]还发现,在高pH值条件下,膨润土对Cs+的吸附以表面配位作用为主。
表面配位模型中的静电相互作用表现为界面区域的不同平面和层数,离子被吸附在不同的位置,常用于探讨黏土矿物与Cs+的吸附机理。采用该模型可得到Cs+在土壤表面和溶液中的吸附形态,通过体系变化可得到相关的反应常数。然而,该模型适用于较理想的均质土壤类型,对于土壤这种典型固液非均相体系,在实验上很难区分化学吸附能和静电库仑能两种不同的相互作用。
3) 离子交换模型
离子交换理论是由Bolt(1982)提出的一种宏观方法,该吸附理论涉及黏土矿物层间阳离子补偿的结构负电荷。通常,可用多个交换位点来校准离子交换量,离子交换模型最初用阳离子交换容量的单点模型来描述,随着研究的深入,逐步发展为多点位离子交换模型[18]。

图3 云母矿物吸附形态和吸附位点示意图Fig.3 Adsorption morphology and site of mica minerals
研究表明,在大多数土壤中,Cs+离子主要吸附在伊利石楔形位点上(图3)[36-37]。由于Cs+几乎完全以一价阳离子的形式存在于溶液中,表面配位模型很难对Cs+的行为做出准确预测,Freundlich吸附模型说明Cs+在伊利石上至少有2个不同的吸附位点[38]。为模拟实验数据,了解黏土矿物对Cs的非线性吸附行为,文献中提出了一系列多点离子交换模型。对于伊利石、蒙脱石和高岭石通常考虑三位点的阳离子交换模型[39-41]。此外,单位点的阳离子交换模型[42-43]和双位点吸附模型[44]也在描述蒙脱石中Cs+的吸附行为研究中得到应用。
Bradbury等[40]建立了一种土壤与Cs的广义吸附模型(GAM),该模型已用于分析不同的实验结果。GAM的基本假设为:(1) Cs+在黏土矿物中的吸附机理为阳离子交换。(2) 存在3种不同的吸附位点类型,它们具有不同吸附容量和亲和性,包括FES、PS和TIIS(由表面Si—O—Si和Al—O—Al断裂形成的边位点)。PS位点浓度与云母类矿物的CEC直接相关(如伊利石为固化体时,其PS位点约占CEC的80%),且吸附形态主要为外层络合态(OSC);TIIS位点浓度低于PS(如伊利石为固化体时,TIIS位点约占CEC的20%)。由层状云母类矿物风化形成的FES位点,其位点浓度相对较小(<10-6mol/g),但其对Cs+的选择性吸附能力远高于PS和TIIS位点,易形成内层络合态(ISC)。对于Cs+,非膨胀性性云母矿物是IS位点,不可利用,而膨胀性云母类矿物可提供高容量的IS位点,当水合Cs+进入IS位点后失去水合氛围,进而诱导层间坍塌至1 nm,此时,位于坍塌层间的Cs+主要以ISC形式存在,活性很低,形成了热力学上更加稳定的结构。
在经典吸附模型的基础上,Fan等[37]提出了Cs在蛭石上吸附行为的四位点优化模型,增加了IS位点的贡献并优化了选择性系数,得到各位点容量、选择性系数和位点百分比参数,验证了GAM改进模型在准确预测沉积物中Cs+分布的可能性。吴涵玉[45]利用四位点的吸附模型,研究了典型云母矿物与放射性Cs的吸附作用机制,连续提取实验和EXAFS光谱澄清了Cs+在风化后云母矿物上的吸附形态,放射性Cs和Th(Ⅳ)/Eu(Ⅲ)复杂体系共存时,2类离子对金云母层间距离的调控具有拮抗作用,2类离子的相对浓度决定云母矿物的层间距离,从而控制云母矿物的风化过程,放射性Cs、Th/Eu调控金云母风化的机理如图4所示。
综上所述,单位点、双位点、三位点、四位点、五位点等离子交换模型已在不同的黏土矿物吸附与解吸行为研究中得到应用。现有离子交换模型与一些实验结果吻合较好,但模型中拟合参数多且不易确定。此外,模型仅适用于校准后的实验数据,在不同土壤环境或自然系统中预测放射性Cs吸附、解吸行为的能力有限。环境中土壤结构类型复杂,单一的离子交换模型难以满足应用需求,因此,需结合环境中土壤类型,开展多种吸附模型融合研究。
4 吸附/解吸行为表征方式及参数
Cs+在黏土矿物中的吸附、解吸作用机制的研究通常是利用光谱法和显微分析技术进行量化分析。
4.1 核磁共振谱分析
核磁共振(NMR)谱的工作原理是原子在磁场中会与特定频率的电磁波发生共振,NMR谱仪通过射频共振测量样品中原子的自旋[46]。NMR谱可用于13C、1H、39K[47]、7Li[47]、23Na[48]、133Cs[49]等阳离子元素分析。其中,133Cs固态NMR分析已用于评价Cs+在黏土矿物中的吸附位点和迁移[50-51]。Kim等[52]采用NMR谱研究了不同条件下Cs+在高岭石和伊利石中的行为,得出Cs+在黏土矿物上的吸附受环境湿度、溶液浓度和Cs+滞留时间的影响,并推断Cs+吸附的2个位点:基面位点和楔形位点。因此,NMR法有助于揭示放射性Cs+在黏土矿物中的吸附位点特性及吸附作用机制。
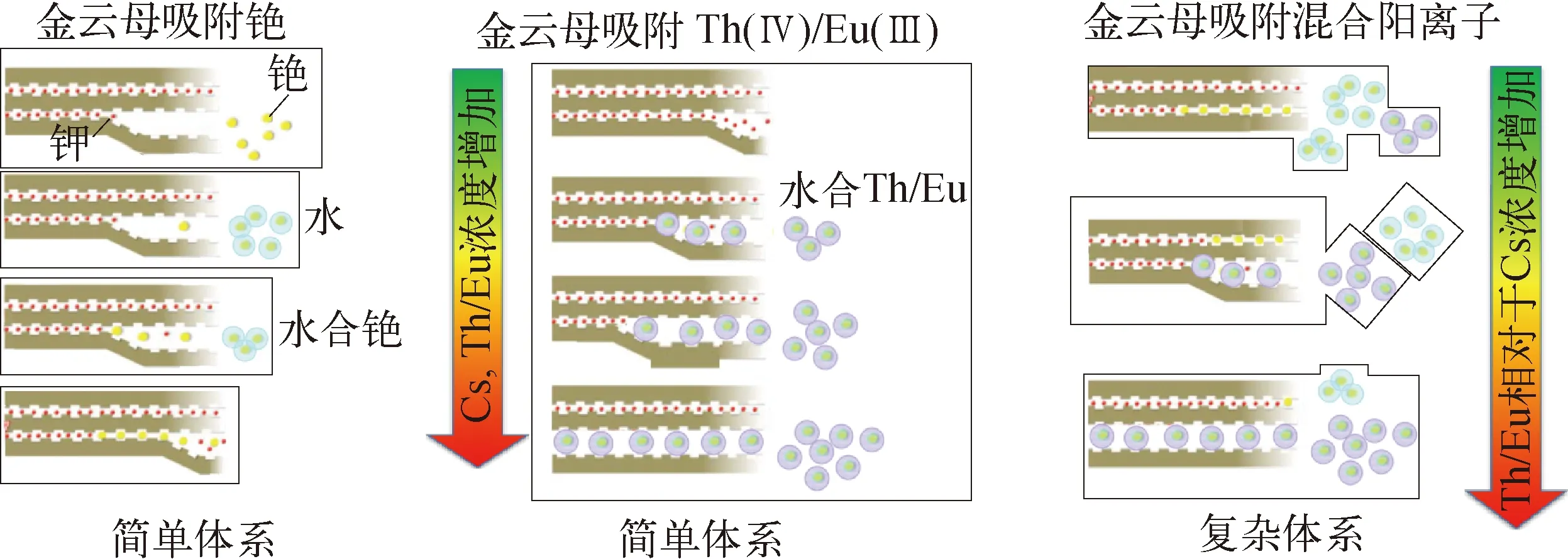
图4 放射性Cs、Th/Eu调控金云母风化的机理Fig.4 Weathering of phlogopite controlled by Cs and Th/Eu
4.2 透射式电子显微镜
透射式电子显微镜(TEM)是研究Cs+与黏土矿物结合形态的有效工具,其原理是真空加速的电子束透过样品,通过电磁场或静电场收集到荧光板上,由于电子通过样品传输,TEM能使样品中的Cs原子成像。黏土矿物中的Cs+被视为黏土矿物结构的一部分。如,Mckinley等[53]观测了Cs+在风化云母矿物上的FES;Fuller等[19]利用TEM拍摄了夹层和FES图像并估算了夹层尺寸。研究发现,借助TEM分析可清晰地观测FES与层间位点尺寸,但在FES和黏土矿物层间仍无法观测到Cs+微粒或离子。此外,有学者利用TEM特别是高分辨透射电镜(HRTEM)对黏土矿物的结构进行了分析,结果表明,层间与FES结构影响了Cs+的吸附[54-55]。Lee等[8]使用能谱透射电子显微镜(TEM-EDX)对Cs+和Ca2+在伊利石中的分布特征进行了研究,得出Cs+主要分布在边缘位点,而Ca2+在伊利石颗粒中随机分布的结论。虽然TEM能直观反映黏土矿物结构,但其观测范围有限,需适当增加覆盖整个黏土颗粒的TEM图像范围。
4.3 扩展X射线吸收精细结构光谱
近年来,扩展X射线吸收精细结构光谱(EXAFS)在环境领域广泛应用,可从分子水平揭示放射性核素与黏土矿物的相互作用机制。EXAFS分析方法对研究Cs+与黏土矿物间的原子键长、原子间距、配位原子种类和数量等微观结构信息作用明显。Fan等[12]揭示了蛭石层间结构塌陷与Cs+吸附形态之间的关系,如图5所示。Cs原子与氧原子发生配位反应,分别形成2个壳层,Cs+被水化第一壳层吸附,形成外层络合(OSC)形态,Cs—O原子间距与Cs+在水溶液中的原子间距相当,键长2.95(蒙脱石)~0.32 nm(伊利石),配位数小于8,为弱吸附形态。相比之下,第2壳层为脱水吸附层,对于内层络合(ISC),Cs—O/Si原子间距变大,键长为0.41~0.43 nm(0.45~0.47nm),配位数小于10,为强吸附形态。通过配位数之比可判定放射性Cs在不同黏土矿物上的赋存形态。
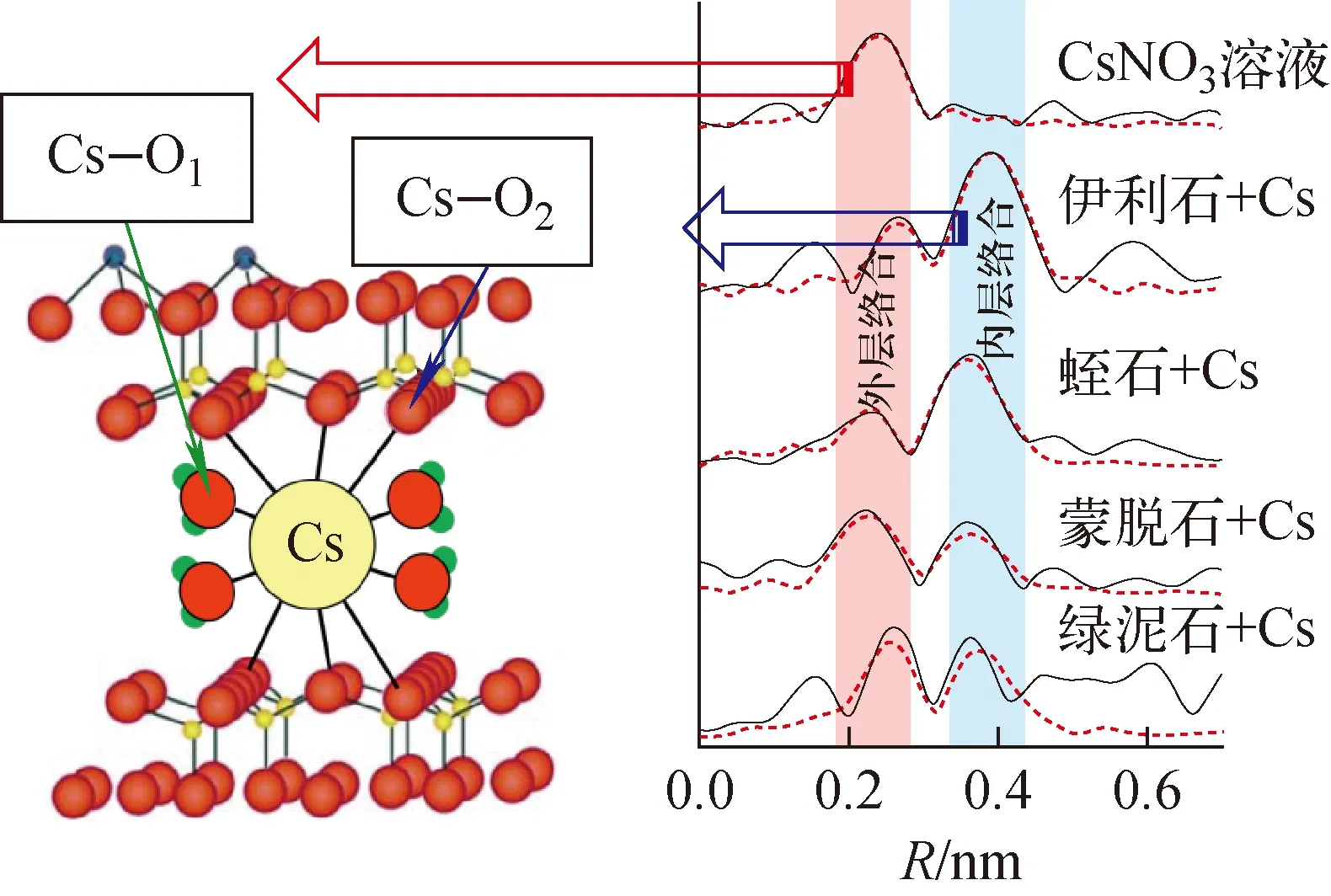
Cs—O1——Cs与土壤水中的氧发生配位;Cs—O2——Cs与四氧化硅中的氧发生配位图5 EXAFS用于Cs+在蛭石矿物上吸附形态分析Fig.5 Speciation analysis of Cs+ adsorbed on vermiculite by EXAFS
Fan等[12]利用EXAFS技术与Tessier连续提取法,研究了Cs+与黏土矿物的作用机制。Cs+在伊利石中易形成ISC内层结合形态,具有强吸附作用而不易解吸;在外层的弱吸附位点形成OSC结合形态,弱吸附作用易解吸。作为中等膨胀性的蛭石黏土矿物,拥有大量的层间吸附位点,当Cs+进入层间结构后失去水合作用,导致蛭石层间晶格结构坍塌,最终形成稳定的晶格层间ISC,如图6所示。有机质存在时,IS和ISC位点被隔离屏蔽,Cs+只能在黏土表面形成OSC。因此,土壤中存在的有机质不利于Cs+的吸附固定。
然而,EXAFS技术也有其局限性,通常需Cs+浓度大于50 mg/kg,自然界中放射性Cs含量极低,有的处于痕量水平,且黏土矿物的FES位点对Cs离子的吸附容量通常低于50 mg/kg,因此,EXAFS技术在低污染核素分析中有一定局限性。
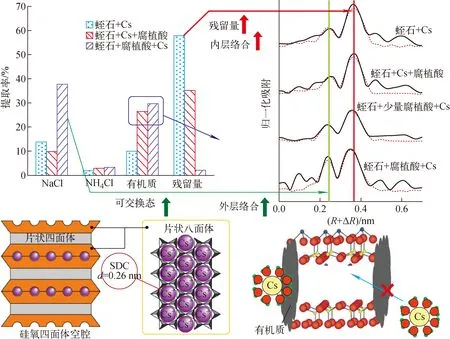
图6 土壤有机质与蛭石颗粒和Cs+的相互作用机制Fig.6 Interaction mechanism of soil organic matter on vermiculite particles and Cs+
4.4 放射自显影
放射性核素显像常用的3种方法分别是胶片自显影、磷屏成像和使用微通道阵列探测器(MICAD)的电子放射自显影(IP)。胶片自显影是一种探测β粒子的方法,其原理是将胶片上的卤化银晶体颗粒中的银原子还原成金属银,使胶片上显示放射性同位素的自显影图像,再利用光学显微镜观测放射性粒子[56]。IP技术是探测土壤中放射性粒子的有效方法。IP成像板网格经放大、立体纤维镜扫描后,可实现颗粒物的定位和微观结构分析,表征土壤微粒在成像板上的精确位置与辐射强度,成像板可重复利用,如图7所示。该方法在土壤、植物组织、鸟类羽毛等样品辐射或放射性Cs分布研究中得到了广泛应用[57]。

图7 土壤颗粒立体显微镜图像Fig.7 Stereomicroscope image of soil particle
土壤中放射性Cs的浓度在10 ppb左右(10-10mol/L),该浓度已接近或低于常规核辐射仪器探测下限,而放射自显影法可解决土壤中微量Cs离子浓度的测量技术瓶颈,通过测量单个黏土矿物颗粒中的辐射强度,评估土壤矿物对137Cs的吸附量。在不同土壤矿物类型、浓度、浸泡时间下,通过IP技术检测到风化黑云母(WB)中137Cs的吸附容量明显高于其他矿物,当137Cs浓度较低或反应时间较短时,仅从WB颗粒中检测到137Cs,如图8所示。实验结果表明:WB在福岛土壤中对放射性Cs有极强的吸附能力,证明了WB对放射性Cs的高富集特征[58]。将含有137Cs的WB颗粒添加至各种电解质溶液中,并通过IP技术可估算浸出前后放射性Cs的解吸量,随着吸附时间的延长,137Cs离子沿WB层间区域迁移扩散,滞留在较为稳定的层间吸附位点,137Cs难以通过阳离子交换作用从层间结构解吸出来[59]。
为测定低放射性活度水平,胶片放射性自显影通常需要很长的胶片曝光时间,胶片自显影用于放射性定量的线性动态范围仅1.5~3个数量级,很难确定样品所需曝光时间,需要2次或多次曝光才能确定胶片线性动态范围内的放射性活度,对低于该阈值的放射性没有反应,且还存在一个饱和点,高于该点的β粒子和γ射线对胶片的光密度没有额外作用,这些缺点限制了该技术的应用。
4.5 不同表征方法的特点与比较
NMR、TEM、EXAFS、IP等表征手段在土壤中放射性Cs吸附与解吸行为表征中均已得到应用,但各有其优缺点(表2),因此,在实际分析中应综合多种表征技术,从宏观与微观2个层面揭示放射性Cs与黏土矿物的作用机制。
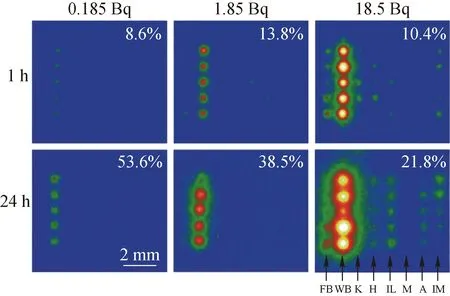
FB,新的黑云母;WB,风化黑云母;K,高岭石;H,埃洛石;IL,伊利石;M,蒙脱石;A,水铝英石;IM,伊毛缟石图8 不同矿物颗粒吸附显影成像Fig.8 Adsorption imaging of different mineral particles

表2 不同表征方法的优缺点Table 2 Advantage and disadvantage of different characterization methods
5 总结与展望
国外针对放射性Cs与黏土矿物的吸附、解吸及迁移等环境化学行为开展了深入研究。通过批式实验开展吸附、解吸宏观研究,针对不同土壤类型,特别是黏土矿物结构对吸附、解吸的作用机制不同,建立了多种吸附模型,其中等温吸附模型、表面配位模型、多位点离子交换模型等得到不同程度的应用,多位点离子交换模型因其可用于解释多数吸附、解吸过程,应用最为广泛。但国内有关放射性Cs的环境行为研究较少,针对我国土壤类型的吸附模型的研究也鲜见报道。目前,放射性污染土壤为多种污染核素混合物,放射性Cs与土壤的吸附模型多为单个模型研究,单一的表征手段很难满足吸附、解吸作用机制的研究需求。因此,建议可从以下几个方面开展研究。
1) 开展土壤中放射性Cs与复杂体系污染核素(238U、90Sr、152Eu、239Pu等)共存时的相互调控作用研究,揭示相互调控机制。
2) 在放射性Cs与黏土矿物微观表征分析技术方面,开展多种表征技术的综合应用,特别是EXAFS、IP等先进表征技术,可从分子水平揭示放射性Cs与黏土矿物的微观作用机制。
3) 现有的吸附模型多是在单一的黏土矿物结构吸附实验基础上提出的,现场土壤类型复杂,多数吸附模型难以满足土壤吸附、解吸行为研究需求,应开展多模型融合研究,建立统一的土壤中放射性Cs吸附解吸行为模型。
4) 结构光谱与EXAFS分析是一种结构平均化的结果,在提取吸附产物中单个吸附构型的结构信息方面还存在一定的困难,可通过分子动力学以分子(原子或离子等)为研究对象[60],考察Cs离子在不同土壤结构中的微观分子运动规律,解决实验手段难以测定表面吸附结构的问题。
5) 针对实际放射性Cs污染土壤中Cs的吸附、解吸行为研究鲜见报道,应开展吸附、解吸模型在实际放射性Cs污染场地的验证及应用研究。
猜你喜欢
环境保护与循环经济(2022年4期)2022-06-30 09:14:08
弹性体(2022年1期)2022-05-11 08:50:46
考试与评价·高二版(2020年1期)2020-09-10 23:34:28
中国特种设备安全(2019年11期)2020-01-16 08:06:14
阅读与作文(英语高中版)(2019年8期)2019-08-27 03:59:11
福建质量管理(2019年15期)2019-03-26 08:10:52
西南石油大学学报(自然科学版)(2018年6期)2018-12-26 01:00:18
橡胶科技(2018年10期)2018-07-21 06:01:58
临床医药文献杂志(电子版)(2017年11期)2017-05-17 04:48:17
中国洗涤用品工业(2015年7期)2015-02-28 19:02:42
