
固相萃取-超高效液相色谱-串联质谱法测定水中16种有机磷农药残留
2021-03-05 09:58:10高振刚曾莎莎曾鸿鹄梁延鹏覃礼堂宋晓红
分析科学学报 2021年1期
高振刚, 曾莎莎, 曾鸿鹄,2,3, 梁延鹏*,2,3, 覃礼堂,2,3, 宋晓红,2,3
(1.桂林理工大学环境科学与工程学院,广西桂林 541004;2.广西环境污染控制理论与技术重点实验室,广西桂林 541004;3.岩溶地区水污染控制与用水安全保障协同创新中心,广西桂林 541004)
有机磷农药(Organophosphorus Pesticides,OPs)以其高效致毒、快速降解的特性在农业生产中被广泛施用[1]。自2015年农业部颁布《到2020年农药使用量零增长行动方案》以来,OPs的施用量虽有所下降,但是敌百虫、敌敌畏、毒死蜱和辛硫磷等10种OPs每年的使用量仍达到1 000~3 0000吨[2]。理论上OPs被施用后,自然条件下能降解为无毒的无机磷,但水环境中OPs的不断检出证明存在OPs污染。因此,有必要建立水中多种OPs的检测方法。
目前,水环境样品常用的前处理方法有液-液萃取(LLE)[3]和固相萃取(SPE)[4],其中SPE效果稳定、溶剂用量少,有利于标准化和自动化。富集OPs常用的SPE小柱有C18柱[5]和HLB柱[6],均为反相SPE柱,适用于非极性至中等极性的化合物,对多数OPs有较好的萃取效果,但对强极性OPs萃取效果较差。而活性炭柱属于无机吸附型SPE柱,适用于强极性有机污染物的富集[7]。OPs的分析多采用气相色谱(GC)法[8],其主要针对易气化且热稳定的物质。OPs种类较多,理化性质差异大,分子质量较大,多具有强极性和热不稳定性,采用液相色谱(LC)法更具优越性[9]。但色谱法仅靠保留时间定性,易出现假阳性结果[10,11],而与质谱联用使色谱和质谱优势得到互补,具有更好的灵敏度与准确度,在极性差异大的农药多残留分析方面具有突出的优势[12]。本研究对固相萃取柱及洗脱液进行优化选择,建立了固相萃取-超高效液相色谱-串联质谱(SPE-UPLC-MS/MS)法同时检测水中16种OPs残留的分析方法,适用于水中多种痕量OPs的检测。
1 实验部分
1.1 仪器与试剂
Waters Xevo TQ-S Micro超高效液相色谱-三重四极杆质谱仪(美国,Waters公司);AQUA Trace ASPE 799型固相萃取仪(日本,岛津公司);双频超声波清洗器;24位氮吹仪;Waters Oasis HLB固相萃取柱(500 mg,6 mL,美国Waters公司);椰子壳活性炭AC固相萃取柱(500 mg,6 mL,德国CNW公司);HF Bond Elut-C18固相萃取柱(500 mg,6 mL,美国Agilent公司)。
16种标样包含敌敌畏、氧乐果、乐果、甲胺磷、乙酰甲胺磷、久效磷、灭线磷、辛硫磷、对硫磷、敌百虫、水胺硫磷、杀扑磷、三唑磷、氯唑磷、马拉硫磷和毒死蜱,纯度≥98%(美国o2si公司),该标样质量浓度为100 μg/mL,介质为甲醇,使用时将其稀释至2 μg/mL制成储备液,避光,-20 ℃冰箱保存。甲醇(色谱纯,纯度>99.9%,德国Merck公司);乙腈、丙酮、二氯甲烷、乙酸乙酯(色谱纯,纯度≥99.9%,美国Fisher公司);甲酸(色谱纯,纯度>98.0%,阿拉丁);其他试剂均为国产优级纯试剂。水为Milli-Q型超纯水仪(美国,Millipore公司)制备的超纯水。
1.2 样品前处理
1.2.1 水样预处理采集1 L地表水水样于棕色玻璃瓶中,按照水样∶甲醇=200∶1的比例在水样中加入甲醇作为稳定剂。水样运回实验室后立即经0.45 μm滤膜过滤,用HCl将样品pH值调节为4,置于4 ℃冰箱中避光保存,于48 h内进行萃取。
1.2.2 固相萃取法将HLB柱与活性炭柱AC串联,先后用10 mL二氯甲烷-丙酮(1∶4,V/V)、10 mL 5 mmol/L NH4Ac甲醇溶液和10 mL超纯水活化串联萃取柱。取500 mL已过滤水样,以3 mL/min流量均匀通过串联萃取柱,用10 mL超纯水淋洗小柱,氮气加压抽真空干燥40 min。两柱拆分,用12 mL二氯甲烷-丙酮(1∶4,V/V)分3次洗脱HLB柱,用12 mL 5 mmol/L NH4Ac甲醇溶液分3次洗脱活性炭AC柱。洗脱液混合后,在35 ℃水浴条件下氮吹至近干,以0.1%甲酸水溶液-甲醇(1∶3,V/V)混合溶液定容至1 mL,使用0.22 μm滤膜过滤后,待测。
1.3 测定方法
1.3.1 色谱条件ACQUITY UPLC BEH C18色谱柱(100 mm×2.1 mm,1.7 μm);流动相A为0.1%甲酸水溶液,流动相B为纯甲醇,梯度洗脱条件:0~2 min,20%B;2~4.5 min,20%~86%B;4.5~8 min,86%~90%B;8~9 min,90%~20%B;9~11 min,20%B。流速为0.3 mL/min;柱温40 ℃;进样量2 μL。
1.3.2 质谱条件电离方式:电喷雾离子源,正离子模式(ESI+);多反应监测(MRM)模式检测;毛细管电压:0.5 kV;离子源温度:150 ℃;脱溶剂温度:600 ℃;脱溶剂气流速:1 000 L/h。
2 结果与讨论
2.1 质谱条件的优化
将100 μg/L标准溶液在combine模式下通过质谱直接进样,对目标物的质谱条件进行优化。根据OPs电离形式,选择各准分子离子[M+H]+或[M+Na]+作为母离子,对毛细管电压、各母离子的锥孔电压和碰撞能量等质谱参数进行优化,优化标准为保证在MRM模式下响应值相对较高且稳定。16种OPs的质谱条件见表1。

表1 目标物的质谱参数

(续表1)
2.2 色谱条件的优化
为提高目标物灵敏度,使色谱峰分离且峰形最佳,实验进样100 μg/L混合标准溶液,对不同水相(含0.01%、0.05%、0.1%甲酸水溶液)结合甲醇作为流动相进行优化。选择甲醇的主要原因是本实验标准溶液均使用甲醇配制,流动相有机溶液与标样溶剂保持一致有利于物质的响应。结果显示随着水相中甲酸含量的降低,极性大的OPs响应有所提高(例如甲胺磷、乙酰甲胺磷等),极性弱的OPs响应值则降低(例如马拉硫磷、毒死蜱)。综合考虑多数物质的响应选择0.1%甲酸水溶液和甲醇作为流动相。图1为16种OPs的总离子流色谱图。

图1 16种有机磷农药的总离子流色谱图Fig.1 Total ion current chromatograms of 16 organophosphorus pesticides
2.3 固相萃取条件的优化
2.3.1 固相萃取柱的选择OPs极性变化范围较大(-0.85≤ lgKow≤ 4.96),在选择SPE小柱时需考虑其适用性。目前水中OPs的萃取多采用C18小柱和HLB小柱,但已有研究表明C18小柱和HLB小柱对强极性OPs的富集效率低于40%甚至为0%[13]。活性炭AC小柱是一种富集强极性化合物的萃取柱,曾用于水中OPs强极性化合物的富集[14]。为实现同时富集多种极性OPs,本实验比较C18和HLB小柱分别串联活性炭AC小柱(串联时AC小柱在下),萃取OPs时目标物的回收率(图2、图3),得出C18小柱串联活性炭AC小柱时16种OPs回收率在3.25%~104.84%之间,平均回收率为60.72%;HLB小柱串联活性炭AC小柱时16种OPs回收率为19.07%~94.23%,平均回收率为65.52%。为保证多数目标物的萃取效率,最终选择HLB小柱串联活性炭AC小柱。
2.3.2 洗脱液的选择根据相似相溶原理,洗脱液必需有足够的选择性只将分析物洗脱,而将吸附力强的杂质保留在柱上[15]。实验研究了乙腈、甲醇、二氯甲烷、丙酮和乙酸乙酯5种溶剂的洗脱效果,结果如图3所示。对于强极性的OPs(甲胺磷、乙酰甲胺磷、氧乐果及久效磷),洗脱效果(平均回收率)为甲醇(68.27%)>乙腈(67.18%)>二氯甲烷(62.49%)>丙酮(50.39%)>乙酸乙酯(35.75%);对于其他中等及弱极性的OPs,洗脱效果(平均回收率)为丙酮(81.08%)>乙腈(77.71%)>二氯甲烷(72.58%)>甲醇(65.69%)>乙酸乙酯(63.74%)。单一溶剂均表现出对中等及弱极性OPs回收率较高,对强极性OPs回收率较低。

图2 不同单一溶剂洗脱串联小柱(C18-AC)对有机磷农药回收率的影响Fig.2 Recoveries of organophosphorus pesticides extracted by tandem columns(C18-AC) with different single elution solutions
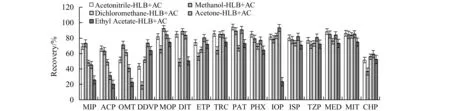
图3 不同单一溶剂洗脱串联小柱(HLB-AC)对有机磷农药回收率的影响Fig.3 Recoveries of organophosphorus pesticides extracted by tandem columns(HLB-AC) with different single elution solutions
由于OPs种类较多,极性差异较大,使用单一溶剂洗脱得不到较好的洗脱效果,故考察5种混合溶剂丙酮-二氯甲烷(4∶1)、丙酮-二氯甲烷(1∶1)、乙酸乙酯-二氯甲烷(1∶1)、丙酮-乙腈(1∶1)和丙酮-甲醇(3∶7)的洗脱能力,结果见图4。可见,对于中等及弱极性的OPs,与单一溶剂相比混合溶剂效果较好,其平均回收率分别为83.47%、82.26%、77.15%、74.03%和63.71%,回收率随着洗脱溶液极性的增大而减小,在其他的研究中也有类似现象[6,16]。对于强极性的OPs,其平均回收率分别为64.66%、55.23%、41.55%、64.00%和69.21%,与单一溶剂洗脱效果相差不大。故选择使用体积比为4∶1的丙酮-二氯甲烷混合溶剂作为HLB小柱的洗脱溶剂。
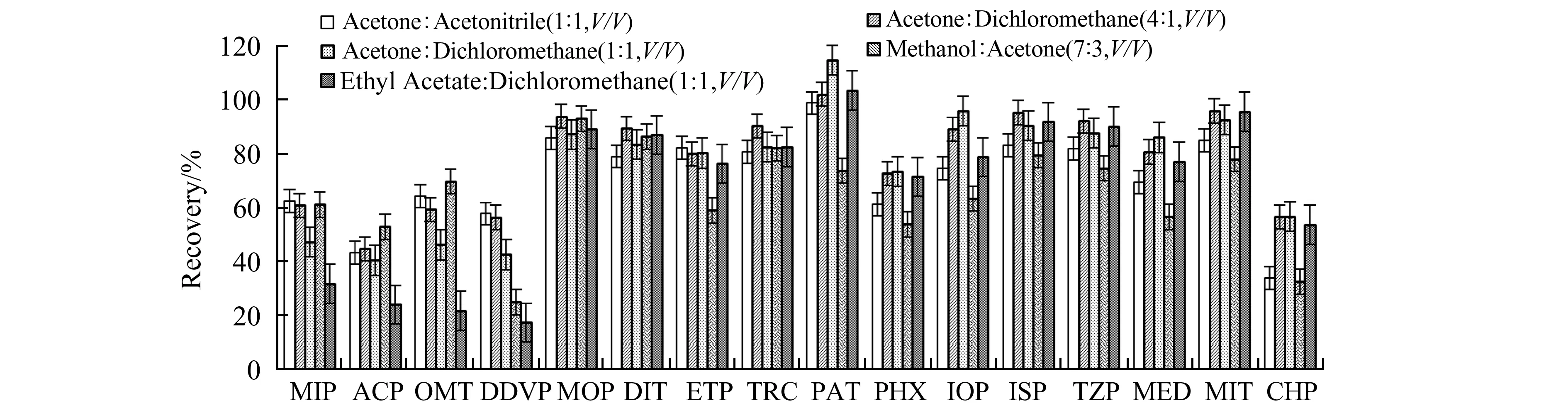
图 4 不同混合溶剂洗脱串联小柱(HLB-AC)对有机磷农药回收率的影响Fig.4 Recoveries of organophosphorus pesticides extracted by tandem columns(HLB-AC) with different mixed elution solutions

图 5 不同溶液洗脱AC小柱对强极性有机磷农药的回收率Fig.5 Recoveries of highly polar organophosphorus pesticides extracted by AC-columns with different elution solutions
因甲醇对强极性有机磷农药萃取效果较好,考察4种溶剂5、10、15 mmol/L NH4Ac甲醇溶液及纯甲醇对活性炭AC小柱的洗脱效果,结果见图5。洗脱液甲醇中加入NH4Ac后,强极性的OPs回收率明显提高。但NH4Ac含量达到15 mmol/L后回收率超出115.00%,与实际情况不符,可能是大量杂质被共洗脱所导致。含5 mmol/L NH4Ac的甲醇平均回收率为102.03%,4种强极性的OPs回收率相对均衡,故选其为活性炭AC小柱的洗脱液。
2.3.3 其他条件优化OPs在碱性条件下易分解,且酸性条件有利于目标物与吸附剂的结合,选择调节水样pH至4;上样流速较慢有利于目标物富集,故选择上样流速为3 mL/min;洗脱液体积采用12 mL,保证能完全将目标物洗脱下来。
2.4 线性范围及检出限和定量限
配制浓度梯度为1、3、5、10、25、50、80、100 μg/L的混合标准溶液,采用优化后的条件进行分析。检测完成后以目标物的浓度(x)为横坐标,目标物的峰面积(y)为纵坐标,绘制回归曲线。结果显示,在1~100 μg/L范围内16种OPs线性良好,相关系数R2≥0.9912。方法检出限以3倍信噪比(S/N=3)计算,定量限以S/N=10计算,结果见表2。检出限为0.004~0.238 ng/L,定量限在0.020~0.794 ng/L之间。

表2 16种有机磷农药的保留时间、线性方程、相关系数(R2)、检出限和定量限
2.5 加标回收率
在500 mL空白水样中加入OPs混合标准溶液,浓度分别为20、100 、200 ng/L,每个浓度水样平行5份,按优化后条件处理并分析水样。回收率及精密度结果见表3。平均回收率在55.5%~109.7%之间,相对标准偏差(RSD)在2.0%~13.9%之间,均在允许偏差内。

表3 16种有机磷农药水样的加标回收率和相对标准偏差(n=5)

(续表3)
2.6 实际样品检测
应用本方法测定了桂林会仙湿地18个点位的地表水,除乐果、灭线磷、杀扑磷和马拉硫磷外,其余12种OPs均有检出,含量为0.038~3.280 ng/L,检出率较高的为毒死蜱(55.5%)>对硫磷(48.1%)>三唑磷(46.3%)>甲胺磷(42.6%)>敌百虫(40.7%)。
3 结论
本研究通过固相萃取-超高效液相色谱-三重四极杆串联质谱法,建立了水中16种有机磷农药的分析方法。在优化的实验条件下,16种目标物具有良好的线性关系,相关系数R2均大于0.991,检出限和定量限范围分别为0.004~0.238 μg/L和0.020~0.794 μg/L。方法精密度高、重现性好、使用试剂环境友好,可同时检测多种污染物,满足水中痕量污染物的分析要求。
猜你喜欢
少年文艺(2022年8期)2022-07-08 10:02:47
时代英语·高一(2019年5期)2019-09-03 02:09:34
上海化工(2018年10期)2018-10-31 01:21:06
中国经济周刊(2017年6期)2017-03-21 00:59:27
读写算·高年级(2016年3期)2016-05-30 01:53:46
电测与仪表(2016年11期)2016-04-11 12:20:42
电源技术(2015年5期)2015-08-22 11:18:28
中国医疗美容(2015年1期)2015-07-12 10:06:18
化工生产与技术(2014年5期)2014-02-27 13:42:02
东南大学学报(自然科学版)(2012年3期)2012-06-28 03:59:00
