
双席夫碱氧钒(IV)配合物抑制多种PTPs活性研究
2021-03-03 06:47王矗宋澳旋李鹏利袁彩霞
山西大学学报(自然科学版) 2021年6期
王矗,宋澳旋,李鹏利,袁彩霞*
(1.山西大学 分子科学研究所,化学生物学与分子工程教育部重点实验室,山西 太原 030006;2.山西大学 化学化工学院,山西 太原 030006)
0 引言
钒是人体必需的微量金属,在生理条件下通常以五价钒形式存在于酸根阴离子(VO43-)或者四价形式存在于钒氧阳离子(VO2+)中[1],其中VO43-在某种程度上类似于磷酸盐,VO2+类似于Mg2+,所以钒化合物不仅具有丰富多彩的化学结构,而且也表现出多种多样的生物活性。目前越来越多的研究发现钒化合物在医药领域有着潜在的应用,尤其是糖尿病治疗方面[2-4]。与无机钒酸盐的糖尿病治疗效果相比,钒配合物在细胞和动物模型实验中表现出更好的生物利用度和更低的毒副作用[5-6]。研究显示配体的螯合能够在一定程度上提高金属离子在生物体内的分布、再吸收能力、稳定性和耐受性,从而极大地影响了钒化合物的生物活性[7-10],最典型的例子是联麦氧钒(BMOV)及其类似物双乙基麦芽酚氧钒配合物(BEOV)[10],该类配合物具有比无机钒酸盐更高的类胰岛素作用,BEOV曾用于二期临床试验,因此钒配合物仍然是抗糖尿病研究的热点。在该领域,目前的主要研究目标之一是找到抗糖尿病活性更高的钒配合物并阐明其机理。
蛋白酪氨酸磷酸酶1B(PTP1B)是抗糖尿病药物作用研究的主要靶点之一[11],但它只是蛋白酪氨酸磷酸酶(PTPs)家族中的一员,在这个一百多名成员的大家族中,几乎每个PTP都与人类疾病密切相关。近期研究表明除PTP1B以外,TCPTP(T细胞蛋白酪氨酸磷酸酶)、SHP-1(含SH2区域的蛋白酪氨酸磷酸酶)和PTP-Meg2(巨核细胞蛋白酪氨酸磷酸酶2)[12-15]也会阻碍胰岛素降糖作用的发挥,即对细胞内胰岛素信号的转导起负向调控作用,所以这些PTPs酶在糖尿病发病机制中也占有重要地位。相应地,能同时有效抑制这四种酶活性的药物将能在更低剂量水平下有效地调控胰岛素受体的去磷酸化水平,使得胰岛素信号正常转导,从而达到在治疗糖尿病的同时降低药物毒性的目的。因此,以多个PTPs为靶点的抑制剂设计是目前抗糖尿病药物开发的另一个热点。
本文研究发现,3个水杨醛缩邻苯二胺衍生物席夫碱氧钒配合物对PTP1B、TCPTP、SHP-1和PTP-Meg2活性均能表现出较高的抑制作用,初步探讨了配合物结构与其活性之间的关系以及该类配合物抑制PTP1B机理,为合理设计基于氧钒配合物的多靶点PTPs高效抑制剂提供了一定的理论基础。
1 实验部分
1.1 试剂与仪器
实验所用化学试剂均为市售分析纯,4种重组PTPs酶(包括 PTP1B、TCPTP、SHP-1和 PTPMeg2)的质粒均由吉林大学生命科学学院邢述课题组提供,本实验室表达提纯[16-17]。配制缓冲溶液用水均为蒸馏水。
实验所用仪器分别为:微量元素分析仪(VARI-EL型),傅里叶红外光谱仪(Shimadiu-FTIR-8300,用溴化钾压片),紫外可见光谱仪(HP-8453),电喷雾质谱仪(Quattro Micro API),全自动酶标仪(Spectra Max 190)。另外,分子模拟工作用Insight II软件在SGI工作站完成。
1.2 配合物的合成
配合物通过一步合成法得到,中间没有分离配体,具体合成步骤为,先将1 mmol邻苯二胺或其衍生物溶于20 mL无水甲醇,搅拌使其完全溶解后,向其中滴加2 mmol水杨醛无水甲醇溶液,继续搅拌并加热回流2 h左右,然后向反应溶液中滴加含有1 mmol硫酸氧钒(0.22 g)的水溶液5 mL,反应溶液用氢氧化钠调pH值到中性,继续回流4 h后停止反应,冷却至室温,过滤,固体粉末依次用蒸馏水、甲醇和乙醚多次洗涤,真空干燥,得到目标产物。
配合物VIVOL1(H2L1=水杨醛缩邻苯二胺席夫碱):产率为0.28 g(70%)。元素分析结果按C20H15.6N2O3.8V (VIVOL1·0.8H2O)计算:Calcd:C60.71%,H3.97%,N7.08%;Found:C 60.75%,H 3.58%,N 7.12%。红外光谱(KBr)ν/cm-1:1 605(C=N),1 318(C—O),980(V=O),626(V—N),484(V—O);紫外可见光谱(DMSO)λmax/nm(ε/103dm3·mol-1·cm-1):263(24.9),317(28.4),407(24.3),592(0.1);电喷雾质谱(VI-VOL1的分子量为381.28):m/z(%),[2(VI-VOL1)+Na+]= 785.08(100%),[3(VIVOL1)+Na+]=1 165.42(24%)。
配合物VIVOL2(H2L2=水杨醛缩-4-甲基邻苯二胺席夫碱):产率为0.24 g(60%)。元素分析结果按 C21H16N2O3V 计算:Calcd:C 63.81%,H 4.08%,N 7.09%;Found:C 63.82%,H 4.12%,N 7.11%。红外光谱(KBr)ν/cm-1:1 602(C=N),1 321(C—O),967(V=O),644(V—N),470(V—O;紫外可见光谱(DMSO)λmax/nm(ε/103dm3·mol-1·cm-1):261(23.8),317(23.9),407(22.4),591(0.1);电喷雾质谱(VIVOL2的分子量为395.30):m/z(%),[2(VIVOL2)+Na+]= 813.08(100%),[3(VIVOL2)+Na+]= 1 207.42(30%)。
配合物VIVOL3(H2L3=水杨醛缩-4-硝基邻苯二胺席夫碱):产率为0.24 g(57%)。元素分析结果按C20H13N3O5V计算:Calcd:C 56.35,H 3.07,N 9.86%;Found:C 55.72,H 3.07,N 9.80%。红外光谱(KBr)ν/cm-1:1 609(C=N),1 326(C—O),968(V=O),644(V-N),488(V—O);紫外可见光谱(DMSO)λmax/nm(ε/103dm3·mol-1·cm-1):263(23.3),321(29.6),427(24.5),617(0.1);电喷雾质谱(VIVOL3的分子量为 395.30):m/z(%),[VIVOL3+H+]= 427.00(54%),[2(VIVOL3)+Na+]=874.50(92%)。
1.3 配合物溶液稳定性实验
将所合成的配合物固体粉末溶于DMSO(二甲基亚砜)溶液,首先用紫外-可见光谱法测定配合物在纯DMSO中72 h内的动力学稳定性,每间隔12 h测定1次,然后用pH为7.2的MOPS[3-(N-吗啉基)丙磺酸]缓冲溶液将配合物稀释到3×10-5mol/L,再次用紫外-可见光谱法测定配合物在MOPS(含质量分数10%DMSO)缓冲溶液中2 h内的动力学稳定性,每隔5 min测定1次。
1.4 PTPs活性抑制实验
PTPs活性抑制实验均以六水合对硝基苯磷酸二钠盐(pNPP)为反应底物,室温下在pH为7.2的MOPS缓冲体系中进行。配制一定浓度的含酶缓冲溶液,PTP1B、SHP-1、PTP-Meg2、TCPTP的浓度分别为 60、200、220和250 nmol/L,然后依次将83 μL的含酶缓冲溶液和10 μL配合物抑制剂加入96孔板,所有系列浓度的配合物都加完之后,向每个孔壁加入2 μL浓度为0.1 mol/L的pNPP,瞬时震荡96孔板启动反应,310 K放置30 min后,用5 μL浓度为2 mol/L的NaOH终止反应,用酶标仪测定反应体系在405 nm处的紫外吸收强度,运用origin程序拟合浓度依赖曲线,从而求得半数抑制浓度IC50值,所有数据都是进行非酶校正实验后的结果,且IC50值实验进行了多次重复。所有配合物溶液都是现配现用。
配合物抑制PTP1B的酶促动力学实验同样是在MOPS缓冲体系中进行,固定酶浓度,分别选取终浓度为0.3,0.5,1,2,4,8 mmol/L的pNPP作为底物,记录在各种底物浓度下不同浓度梯度的抑制剂反应初速率的变化情况,运用Excel程序根据Lineveaver-Bruk方程拟合并处理这些动力学实验数据[18],分析配合物的抑制类型并求得其抑制常数。
1.5 配合物与PTP1B的分子对接实验
氧钒配合物与PTP1B相互作用的分子对接实验使用Insight II 2000软件包在SGI工作站上完成,配合物的初始结构由密度泛函理论计算所得(在Gaussian 03软件包中完成[19]),PTP1B晶体结构从蛋白数据库下载[20],运用Insight II程序中的BIOPOLYMER模块添加氢原子,由于Insight II 2000的各个力场中只有ESFF力场含有钒原子的参数,因此所有含配合物的体系都在ESFF力场下进行能量最小化和优化计算。
2 结果与讨论
2.1 配合物结构及其表征
配合物的合成路线如图1所示,结构通过红外光谱、紫外光谱、元素分析、电喷雾质谱和电子顺磁共振谱进行表征,部分数据如实验1.2所示。

图1 双席夫碱氧钒(IV)配合物的合成路线图Fig.1 Synthesis route of oxovanadium complexes with bis-Schiff base ligands
从配合物的红外光谱数据可以看出,980、967和968 cm-1处的强吸收峰分别是配合物VIVOL1、VIVOL2和VIVOL3的V=O特征峰[21-22],1 602 cm-1~1 609 cm-1处的强吸收峰可以归属为配合物中C=N的伸缩振动峰[1],1 313 cm-1~1 326 cm-1处的吸收峰为配合物中酚羟基的C—O伸缩振动峰,配位键V—N和V—O的伸缩振动峰出现在626 cm-1~649 cm-1和470 cm-1~488 cm-1范围内[16];配合物紫外光谱数据显示除了紫外区的吸收峰外,在400 nm以上的可见区也出现明显的吸收峰,这可能归因于配体与金属之间的荷移跃迁和金属离子的d-d跃迁。红外和紫外光谱分析都表明氧钒配合物的形成。
在红外和紫外光谱分析的基础上,我们采用元素分析方法测定了各个配合物中碳、氢、氮比例及其百分含量,结果表明实测值和理论值符合很好,初步判断所合成配合物是目标配合物,同时我们测定了各个配合物分别溶解于甲醇中的阳离子电喷雾质谱图(如图2a-c),在图谱中能观察到相应配合物的分子离子峰、二聚体分子离子峰或者三聚体分子离子峰,该结果进一步佐证了目标配合物的结构。
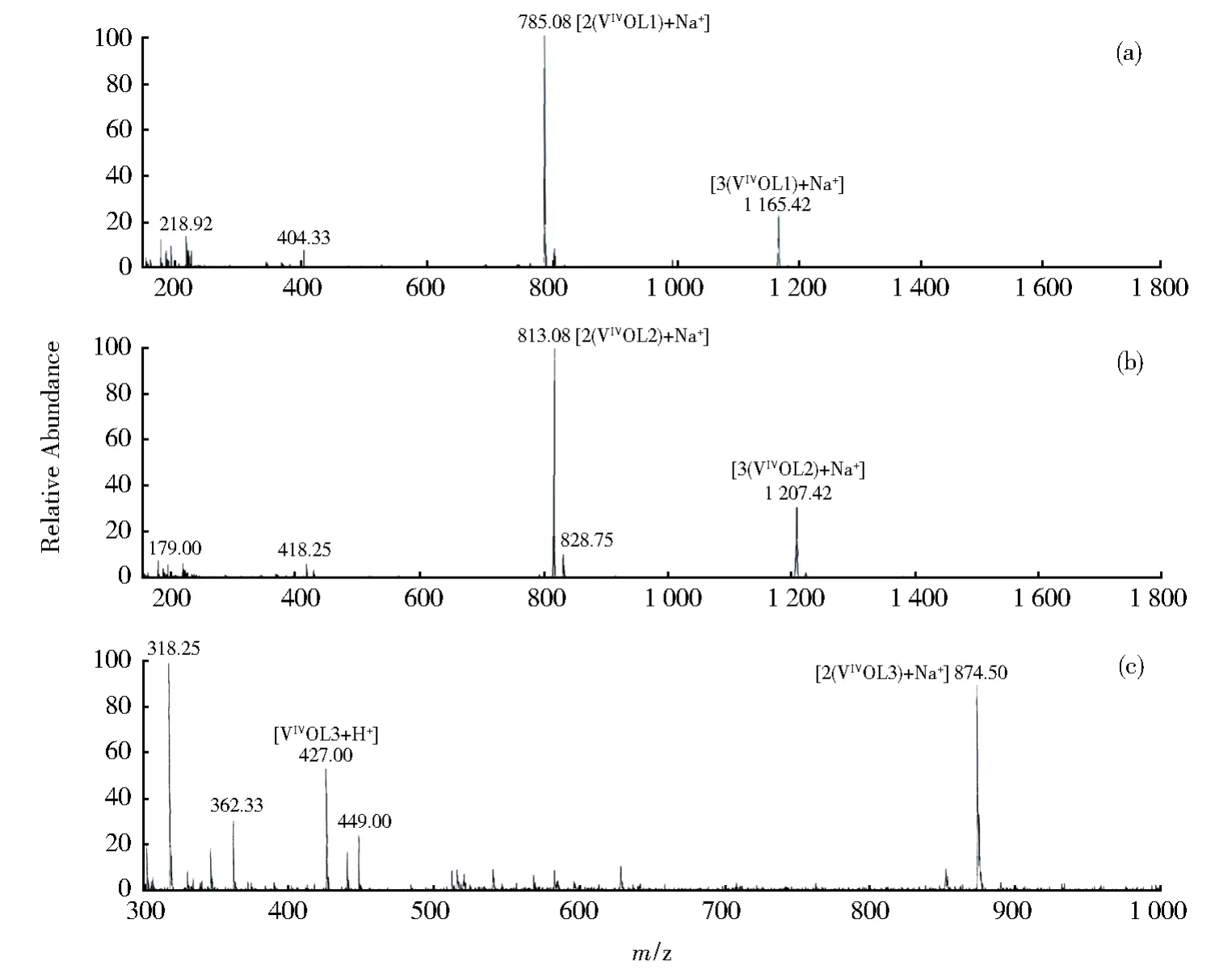
图2 配合物VIVOL1(a),VIVOL2(b)和VIVOL3(c)在甲醇溶液中的电喷雾质谱正离子峰Fig.2 Positive ESI-MS spectra of VIVOL1(a),VIVOL2(b)and VIVOL3(c)in methanol solution
为了更好地评估该类氧钒配合物的价态及其结构,我们以配合物VIVOL1为代表,测定了该配合物在DMSO溶液中的电子顺磁共振谱,如图3所示,配合物呈现出一个四价钒化合物图谱特征的多重超精细结构,表明配合物中钒为正四价,也进一步说明配合物的五配位构型。
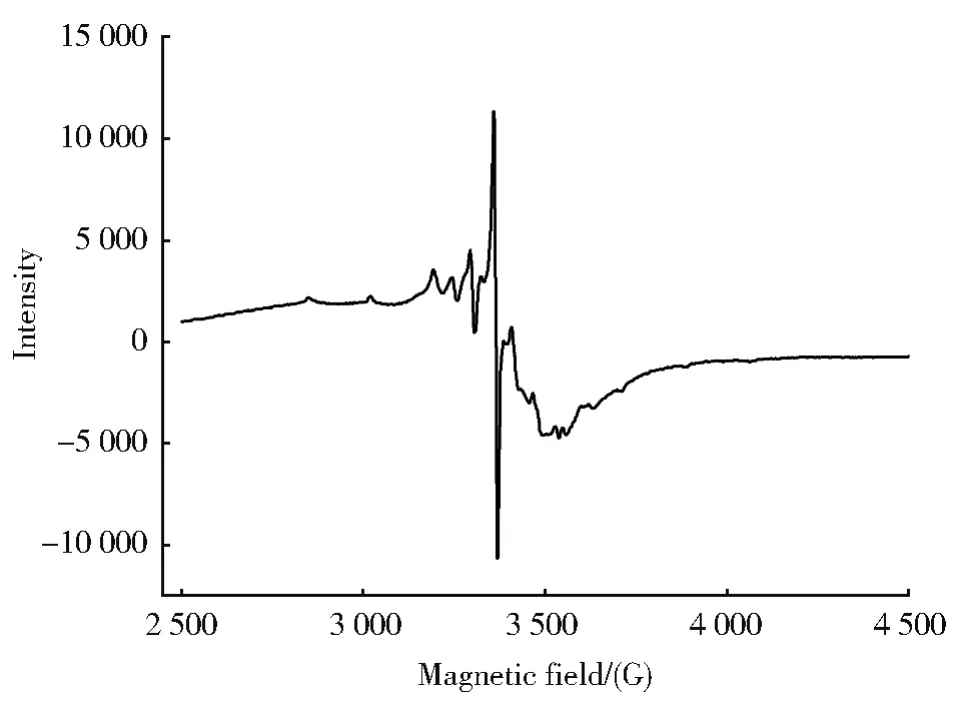
图3 110 K时配合物VIVOL1在DMSO溶液中的电子顺磁共振谱Fig.3 EPR spectrum of VIVOL1 in DMSO solution at 110 K
2.2 配合物溶液稳定性分析
该类氧钒(IV)配合物易溶于DMSO,能溶于甲醇,但是在水中溶解度相对较低,所以其PTPs抑制活性实验都在含10%DMSO的MOPS(pH=7.2)缓冲体系中进行,考虑到配合物溶液稳定性与其生物活性之间的密切相关性,在研究配合物抑制PTPs活性之前,我们首先运用紫外-可见光谱法测定了配合物分别在纯DMSO中放置3 d和在MOPS缓冲溶液(含有10%DMSO,pH=7.2)中放置2 h的稳定性,结果表明,所有配合物在DMSO溶液中都能够稳定存在,在含10%DMSO的MOPS缓冲溶液中,配合物VIVOL1和VIVOL2的紫外吸收峰随时间的延长不发生变化(如图4a和b),但是配合物VIVOL3在缓冲溶液中紫外吸收峰随时间的延长整体有些红移(如图4c),强度略有增加,我们推测可能是该配合物中的硝基在溶液中容易形成氢键所致,实验结果表明配合物VIVOL1和VIVOL2在缓冲溶液中的稳定性明显强于配合物VIVOL3,前两种配合物后续的PTPs抑制实验中主要活性组分就是配合物自身结构,最后一种配合物在缓冲溶液中可能会出现氢键结构。
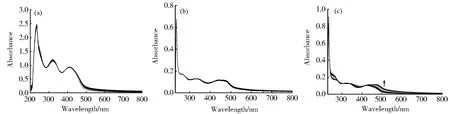
图4 配合物VIVOL1(a),VIVOL2(b)和VIVOL3(c)在310 K、pH为7.2的MOPS缓冲溶液中2 h内的紫外光谱图,每间隔10 min测定1次Fig.4 UV-vis spectra of complexes VIVOL1(a),VIVOL2(b)and VIVOL3(c)recorded at 310 K in MOPS buffer(pH=7.2)over 2 h with 10 min intervals
2.3 配合物对各种PTPs活性的抑制作用
以pNPP为底物,测定了该类氧钒配合物抑制四种蛋白酪氨酸磷酸酶PTP1B、TCPTP、SHP-1和PTP-Meg2活性的IC50值(如表1所示),从表中数据可以看出氧钒配合物抑制四种酶活性的IC50值范围是 0.015 μmol/L~0.116 μmol/L,其中 VIVOL1抑制TCPTP和PTP-Meg2的IC50值分别为0.015 μmol/L和0.016 μmol/L,VIVOL3抑制PTP1B活性的IC50值为0.015 μmol/L,如此低的IC50值,体现出配合物对这几种酶强的抑制能力,但是类似结构的配合物VIVOL2抑制TCPTP活性的IC50(0.116 μmol/L)却大于另外2个配合物,虽然配合物结构类似,但是配合物中取代基的改变会影响其对各种酶的抑制能力。为了更好地理解不同配合物结构对PTPs抑制活性的影响,进一步比较了配合物和硫酸氧钒对酶活性的抑制作用,结果表明硫酸氧钒抑制PTP1B活性的IC50值约是配合物VIVOL1和VIVOL2的两倍和配合物VIVOL3的5倍,抑制TCPTP活性的IC50值约是配合物VIVOL1的6倍,由此可见,3个配合物对PTP1B的抑制能力明显强于硫酸氧钒,其中配合物VIVOL3和VIVOL1几乎是目前基于氧钒配合物的最强的PTP1B和TCPTP抑制剂,而且该类配合物不仅对PTP1B和TCPTP有强的抑制作用,也能够有效抑制PTP-Meg2和SHP-1活性,显示出很好的多靶点PTPs抑制能力。
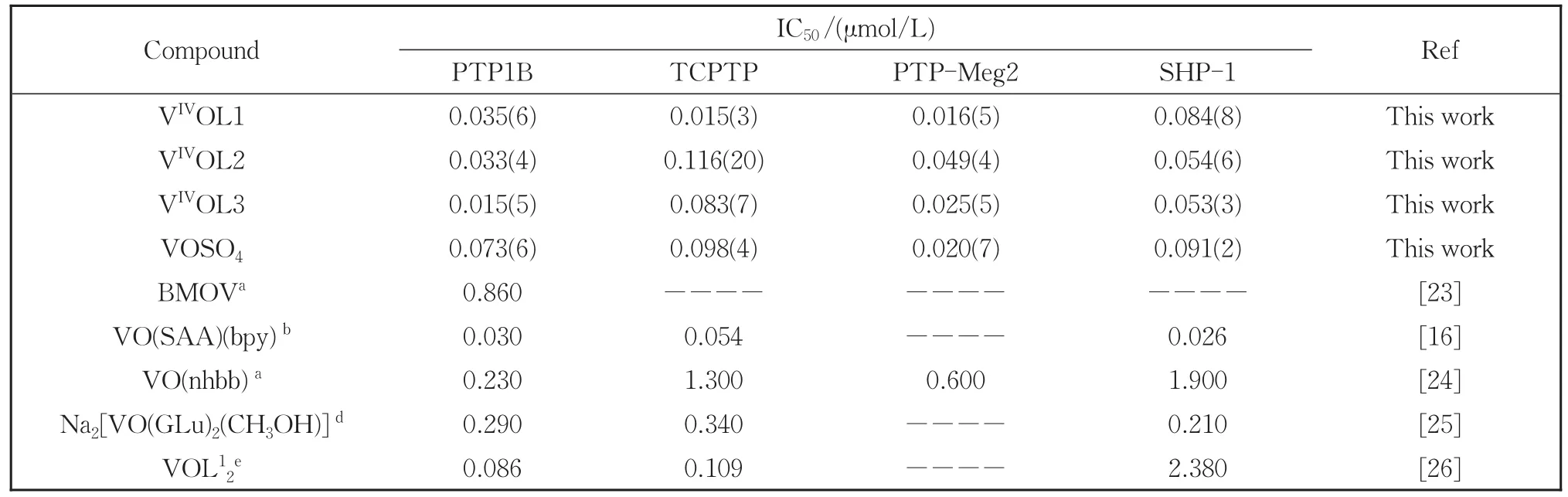
表1 氧钒配合物抑制PTP1B活性的IC50值Table 1 IC50values of the oxovanadium(IV)complexes inhibitors with PTP1B
从表1中数据也可以看出,与部分已报道的氧钒配合物 PTPs抑制剂相比[16,23-26],该类配合物对PTP1B的抑制作用要明显强于BMOV、三齿配位的席夫碱氧钒配合物VO(nhbb)以及谷氨酸氧钒配合物 Na2[VO(GLu)2(CH3OH)],略强于二甲双胍氧钒配合物VOL12,而与混配的水杨醛席夫碱氧钒配合物VO(SAA)(bpy)相似,很显然不同空间结构的氧钒(IV)配合物,对PTP1B活性的抑制能力不同。由此可见,配合物的空间结构对其抑制酶的活性和机理有明显影响。我们推测配合物对各种PTP酶抑制能力的强弱可能与配合物结构和酶本身的结构有关,配合物结构的合理设计将有助于发展具有高效抑制能力的多靶点氧钒配合物抑制剂。此外,众所周知BMOV是较早被确认具有降血糖作用的氧钒配合物,其与PTP1B相互作用机理研究也最为透彻,研究显示BMOV以慢结合方式混合竞争性抑制PTP1B活性,本文所研究的氧钒配合物对PTP1B的抑制作用明显强于BMOV,这可能是由于不同结构的配合物对PTP1B抑制的方式不同所导致的结果,为此我们研究了该类配合物抑制PTP1B活性的作用方式。
2.4 配合物VIVOL1与PTP1B的作用方式研究
选择VVIOL1为代表,首先通过动力学方法分别研究了1 μmol/L和0.1 μmol/L的配合物VVIOL1在不同的作用时间内对PTP1B活性的抑制作用(如图5所示),由图可以看出,当配合物浓度为1 μmol/L时,5 min之内配合物就能够完全抑制PTP1B活性,当配合物浓度为0.1 μmol/L时,20 min内配合物能够抑制90%以上的PTP1B活性,而文献中BMOV对PTP1B反应活性的抑制动力学结果显示[23],当 BMOV 浓度为 37.5 μmol/L 时,60 min内配合物才能够抑制约50%的PTP1B活性,可以看出配合物对PTP1B活性抑制的能力明显强于BMOV,抑制速度也比BMOV快很多,是一种高效快速的PTP1B抑制剂。
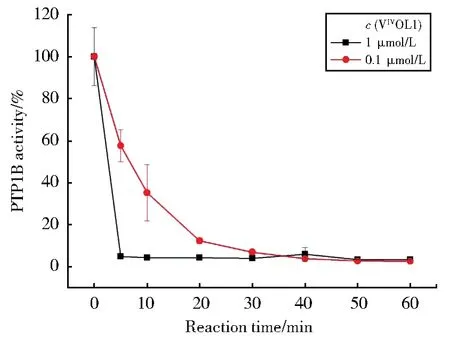
图5 不同浓度的配合物随着作用时间延长对PTP1B活性抑制的影响动力学图Fig.5 Kinetic plot of PTP1B inhibition by different concentrations of VVIOL1 with the extension of the reaction time
在酶动力学实验中,我们根据图5的实验结果在0.1 μmol/L~1 μmol/L范围内选取5个配合物抑制剂浓度,同时在每个抑制剂浓度一定的条件下,从低到高改变底物的浓度,测定随着底物浓度的增加初始反应速率的变化值,用初始反应速率与底物浓度的倒数作图,可以得到如图6所示的双倒数曲线,从图中可以看出,这些曲线都交于Y轴上的一点,表明配合物能够竞争性抑制PTP1B活性,与BMOV的抑制类型不同,进一步以每条双倒数曲线的斜率值对配合物浓度作图可得一条直线(如图7),从该直线的斜率即可求得配合物的抑制常数为0.03 μmol/L。
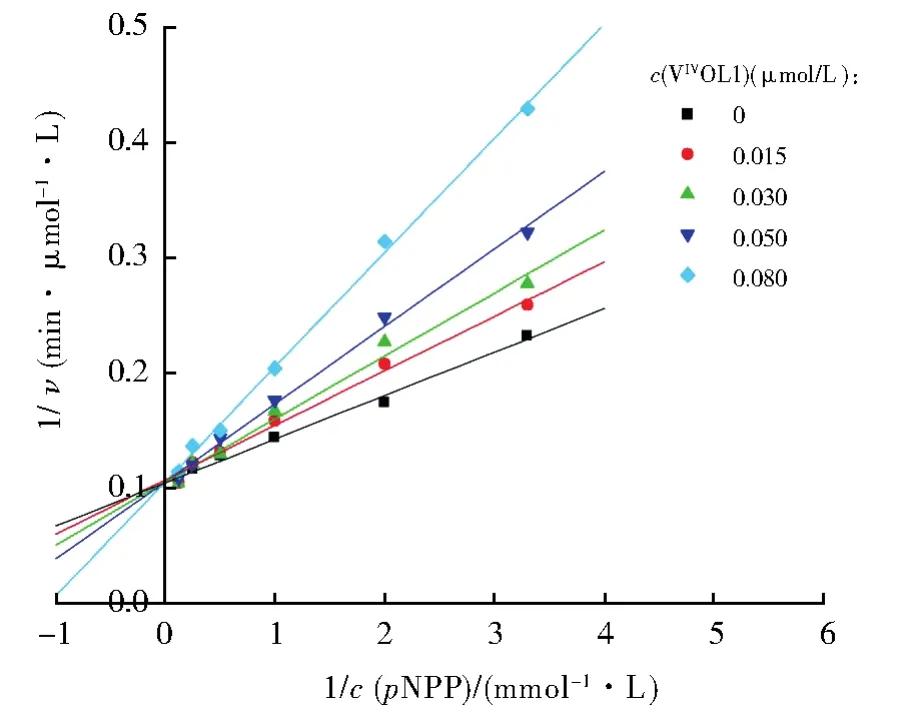
图6 配合物VIVOL1抑制PTP1B活性的双倒数曲线图Fig.6 Lineweaver-Burk plot of 1/ν(min·μmol-1·L-1)versus 1/c(pNPP)(mmol-1·L)at five fixed concentrations of VIVOL1
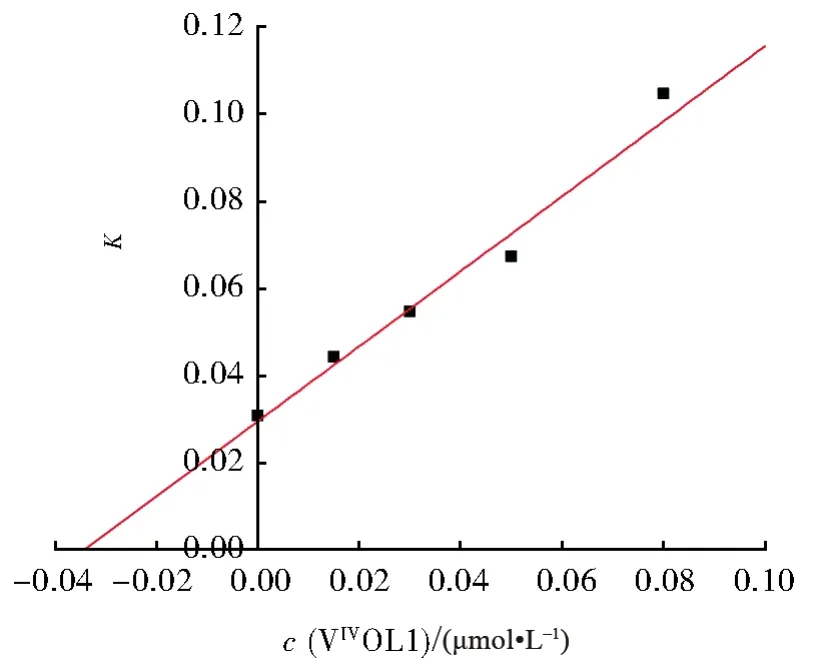
图7 配合物VIVOL1对PTP1B活性的抑制常数测定图Fig.7 Plot of apparent Michaelis constant(K)versus c(VIVOL1)to determine the inhibition constant against PTP1B
2.5 配合物VIVOL1抑制PTP1B活性的作用机理推测
从酶动力学实验可知,配合物VIVOL1以竞争模式抑制PTP1B活性,这种抑制类型是指抑制剂和底物竞争结合酶的活性位点,PTP1B的活性位点是由包含Cys-215在内的八种氨基酸残基构成的刚性环状口袋,为了直观地分析配合物与PTP1B活性位点的作用方式,我们用分子对接的方法模拟了二者在活性位点的作用(如图8所示),从分子对接结果可以看出配合物VIVOL1结构能够进入PTP1B的催化域口袋,其钒氧端靠近活性位点的Cys-215上的硫原子,且配合物中的氧原子易与活性区域的氨基酸残基形成氢键,这些都有利于配合物与酶的竞争性结合,从而抑制酶的活性。
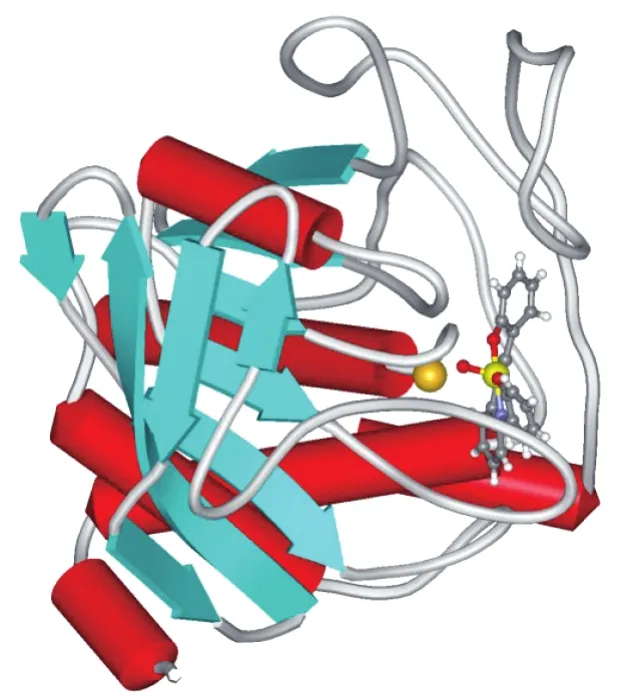
图8 配合物VIVOL1与PTP1B活性位点作用的分子对接图Fig.8 Molecular docking model for PTP1B active poket site of the complex VIVOL1(the sulfur atom of the active site residue Cys215 is highlighted as a yellow ball)
3 结论
本文研究了3个水杨醛缩邻苯二胺衍生物双席夫碱氧钒(IV)配合物对PTP1B、TCPTP、SHP-1和PTP-Meg2活性的抑制作用,结果显示该类配合物都能够有效抑制四种酶活性,且3个配合物对各种酶抑制能力不尽相同,其中值得一提的是,配合物VIVOL3抑制PTP1B活性的IC50值为0.015 μmol/L,该数值显著低于经典氧钒配合物BMOV抑制剂的IC50值,几乎是目前PTP1B抑制活性最高的二氧钒配合物,而且该配合物对PTP1B活性的抑制类型为竞争型,不同于BMOV对PTP1B活性的混合抑制类型,意味着配合物与BMOV具有不同的PTP1B抑制机理。此外,配合物对与胰岛素受体负调控相关的四种PTPs酶活性均能够有效抑制,表现出明显的多靶点抑制作用,具有潜在的抗糖尿病活性。本文的工作为基于二氧钒配合物的多靶点抗糖尿病药物开发提供了新思路。
猜你喜欢
现代临床医学(2022年3期)2022-06-06
体育科技文献通报(2022年3期)2022-05-23
中国典型病例大全(2022年11期)2022-05-13
健康体检与管理(2022年4期)2022-05-13
安徽医科大学学报(2022年4期)2022-05-12
医学概论(2022年4期)2022-04-24
世界中医药(2021年22期)2021-01-03
教育周报·教育论坛(2020年3期)2020-10-21
科技资讯(2018年16期)2018-10-26
科技信息·下旬刊(2018年8期)2018-10-21
