
锕系元素的非寻常氧化态化学Ⅱ.分子配合物
2021-03-02 00:54苏一茂黄闻亮
核化学与放射化学 2021年1期
邓 翀,苏一茂,黄闻亮
北京分子科学国家研究中心,稀土材料化学及应用国家重点实验室,放射化学与辐射化学重点学科实验室,化学与分子工程学院,北京大学,北京 100871
锕系元素是89号元素锕到103号元素铹共15个金属元素的总称。传统上,前七个元素(锕至镅)统称为前锕系元素,而后八个元素(锔至铹)统称为后锕系元素。近年来,非水相的锕系元素配位化学与金属有机化学得到了迅猛的发展[1-2]。譬如,锕系金属-锕系金属多重键[3-5]、铀氮三键[6-8]、铀-芳烃δ反馈键[9-11]等特殊共价体系的理论预测与合成表征,增进了人们对5f轨道参与成键的认识。另一方面,许多高活性锕系金属配合物与小分子或离子的反应,实现了新的配位或活化模式[12-14]。Haber等最早在研究合成氨工艺时,发现铀触媒表现出比铁触媒更高的催化活性[15]。这启发了一系列基于铀配合物的固氮反应研究[16-19],而这些研究又为理解金属铀促进的Haber-Bosch过程提供了新的观点。近年来,锕系金属中心参与的异相催化反应也取得了新的进展[20],而许多锕系金属配合物的均相催化性能和机理也已得到研究[21-26]。尽管目前锕系金属配合物催化的转化效率尚不理想,但这些研究无疑为分子基锕系金属催化剂的发展奠定了基础。此外,新一代的海水提铀工艺[27-30]、优化PUREX等核燃料湿法后处理流程[31-33]等领域,也亟需发展对锕系元素有更高特异选择性的配体体系。
对于非寻常氧化态的锕系元素化学的研究不仅能拓展科学的边界,也有着广泛的应用前景。比如,理论预测表明,许多低价体系有可能实现锕系-锕系金属键配合物的合成[34-36]。又如,将Bk3+氧化成Bk4+有助于锫的分离[37]。在系列综述第一部分中,重点介绍了气相、固相和水溶液中的锕系元素非寻常氧化态化学[38]。作为系列综述的第二部分,本文将着重介绍锕系元素非寻常氧化态化学在分子配合物领域中的发展历史与最新进展。对于研究较多的元素,如钍、铀等,将单独进行介绍,而其他锕系元素则分为前锕系与后锕系两类分别介绍。最后,将对这一领域进行总结和展望。
1 锕系元素的非寻常氧化态
不同于稀土金属收缩于离子核内部的4f轨道,锕系金属的5f轨道基本不被6s和6p亚层所屏蔽,而5f轨道比4f轨道在原子核外也更加扩展[2]。2000年以来,对相似体系中镧系/锕系金属配合物的对比研究,也证实了5f轨道较4f轨道能更好地参与形成共价键[39-41]。不过,近年来也有一些研究得到了不同的结论:比如理论计算表明,M(OC6H5)4体系中M—O键共价性顺序按M=U>Ce>Th顺序递降[42];Gregson[43]和Cheisson[44]等通过实验发现,在三脚架型氮氧配体体系与卡宾二亚胺体系中,铈与配体成键的共价性强于钍与配体成键的共价性。这反映出f轨道参与成键的复杂性,而深入理解f轨道的成键作用还需要更多系统性的合成表征与理论计算研究。其中,针对那些处在“传统价态”之外的锕系金属化合物开展实验和计算研究,能够显著拓宽对于5f轨道的成键及其共价性的认识。
延续文献[38]对锕系元素的“非寻常氧化态”的定义,将Ac(Ⅱ)、Th(Ⅱ)/(Ⅲ)、Pa(Ⅱ)/Pa(Ⅲ)、U(Ⅱ)、Np(Ⅱ)、Pu(Ⅱ)、Am(Ⅶ)、M(Ⅱ)/M(Ⅳ)(M=Cm、Bk、Cf、Es、Fm、Md、No、Lr)等视为锕系元素的“非寻常氧化态”,并在本综述中予以讨论。
2 分子配合物
2.1 钍
(1) Th(Ⅲ)
1974年,Baumgärtner课题组发现,在萘存在的条件下,金属钠在四氢呋喃(THF)中可以将Cp3ThCl(Cp=C5H5)还原,从而得到首例Th(Ⅲ)分子配合物Cp3Th[45]。Cp3Th是一种紫色的、具有弱顺磁性的物质。随后,Kalina等[46]提出了一种制备Th(Ⅲ)茂配合物的新方法:用汞灯照射Cp3Th(i-Pr)(i-Pr:异丙基)或(C5H4Me)3Th(i-Pr)的苯溶液可以得到(C5H4R)3Th(R=H或Me),同时伴有丙烷、丙烯的生成。这种方法同样适用于Th(Ⅲ)茚配合物Th(C9H7)3的制备[47]。
1986年,Blake等[48]报道了首例Th(Ⅲ)配合物的晶体结构。在甲苯中,用过量的Na/K合金处理Cp″2ThCl2(Cp″=1,3-C5H3(SiMe3)2)可以得到暗蓝色的Cp″3Th。单晶X射线衍射(SXRD)显示,Cp″3Th的三个η5配位的茂环对称地分布在Th(Ⅲ)周围(图1)[48]。Kot[49]、Edelstein等[50]利用变温电子顺磁共振(EPR)实验说明,Cp″3Th具有6d1的基态电子构型。Bursten等[51]针对模型配合物Cp3Th的理论计算得到了相同结论。在计算中考虑相对论效应,将使得5f轨道能量上升,而6dσ轨道能量稍微下降,从而在能量上有利于6d1的电子构型[52]。
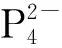
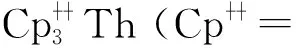
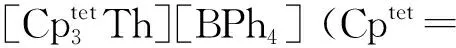
Th(Ⅲ)与环辛四烯(COT)类配体形成的配合物也有报道。1999年,Parry等[65]在DME中用钾镜还原Th(COTTBS2)2(COTTBS2=1,4-C8H6-(SiMe2t-Bu)2),得到了具有三明治结构单元的Th(Ⅲ)配合物,(COTTBS2)Th(μ-COTTBS2)K(DME)2。EPR谱学研究表明,其中Th(Ⅲ)具有6d1的基态电子构型。在该配合物的结构中,钍—碳键的平均距离为2.78 Å(1 Å=0.1 nm),略大于U(Ⅲ)对应物和前体Th(Ⅳ)配合物中的金属—碳键的键长[65]。
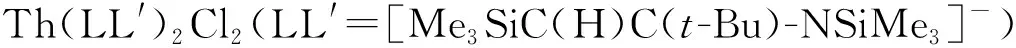
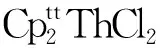
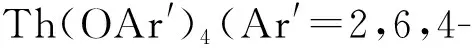
(2) Th(Ⅱ)
1986年,Blake等[48]发现,在用Na/K合金与Cp″3ThCl反应合成Cp″3Th时,有单质钍生成。结合其他实验证据,他们认为,整个反应过程依次经历了Cp″2ThⅢCl→Cp″2ThⅡ→Th(0)+Th(Ⅳ),其中Cp″2ThⅡ发生歧化反应产生了单质钍。随后,他们进一步发现,在室温下,向Cp″3ThCl的THF溶液中加入Na/K合金可以得到一种暗蓝色溶液,该溶液在1 h后会转化为暗绿色。EPR和核磁共振(NMR)测试均表明,这一暗绿色溶液中不含顺磁性物种。据此,他们认为体系中可能含有自旋配对的具有6d2基态电子构型的Th(Ⅱ)配合物,其组成可能为[K(THF)x][Cp″3Th]或Cp″2Th(THF)y。关于它的反应性研究表明,假想的暗绿色Th(Ⅱ)溶液能与叔丁基溴作用生成Cp″2ThBr2[59]。这是最早发现的Th(Ⅱ)可能在分子配合物中存在的证据。
图配合物的合成[74]
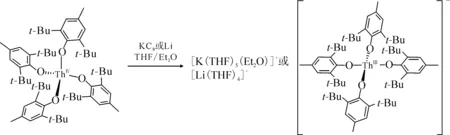
2015年,Langeslay等[60]报道了首例经过结构表征的Th(Ⅱ)分子配合物。他们从Blake等[59]前期的工作中得到启发,通过引入穴醚或冠醚作为阳离子螯合剂,用KC8在THF溶液中还原Cp″3Th,终于分离得到了深色的Th(Ⅱ)配合物[K(crypt)][Cp″3Th] (crypt=穴醚[2.2.2])和[K(18-c-6)(THF)2][Cp″3Th](图3)。比较单晶结构数据发现,[Cp″3Th]-的晶体结构与Cp″3Th十分相似。其溶液多核(1H、13C、29Si)NMR谱均具有抗磁特征,而磁学性质测试也证实其表现为抗磁性。DFT计算表明,[Cp″3Th]-的基态电子构型为自旋配对的(6dz2)2单线态,不同于气态Th2+自由离子的5f16d1的基态电子构型[85]。这凸显了Cp″3体系的平面三角形配位场为6dz2轨道提供的特殊稳定性。值得一提的是,这两个Th(Ⅱ)配合物是首例含有自旋配对的nd2电子构型的非d区金属配合物。
[Cp″3Th]-可以与COT发生两电子转移反应,生成Th(Ⅳ)配合物Cp″2Th(COT)[60]。在[Et3NH][BPh4]的作用下,[Cp″3Th]-被转化为两种含钍的配合物:Th(Ⅳ)配合物Cp″3ThH与Th(Ⅲ)配合物Cp″3Th。此外,[Cp″3Th]-能直接还原氢气,无论是在固相还是在THF溶液中,均可以得到Th(Ⅲ)/Th(Ⅳ)的混价配合物[K(18-c-6)(Et2O)][Cp″2ThⅢ(μ-H)3ThⅣ(H)Cp″2]以及Cp″3Th[86]。最近,Moehring等[87]通过原位EPR监测研究,比较了低价稀土与锕系金属类似物还原能力的相对强弱。他们发现,在THF中,[K(crypt)]-[Cp″3Th]或[K(crypt)][Cp″3U]可以将三价稀土配合物Cp″3M(M=Y、La)还原至二价的 [Cp″3M]-。而逆向反应亦可发生,即二价稀土金属配合物[Cp″3M]-(M=Y、La)也能够还原Cp″3Th或Cp″3U至二价。这一结果表明,在这些Cp″3体系中,二价锕系金属Th(Ⅱ)、U(Ⅱ)与二价镧系金属Y(Ⅱ)、La(Ⅱ)具有相近的还原能力[87]。
具有6d2基态电子构型的Th(Ⅱ)配合物的反应性在未来值得进一步探索。Di Santo等[88]关于Th2+或U2+与小分子烯烃、炔烃的气相反应的研究表明,Th2+的6d2激发态(几乎与5f16d1基态简并)在活化C—H、C—C键过程中起到了至关重要的作用,并且表现出与U2+反应性的显著差别。
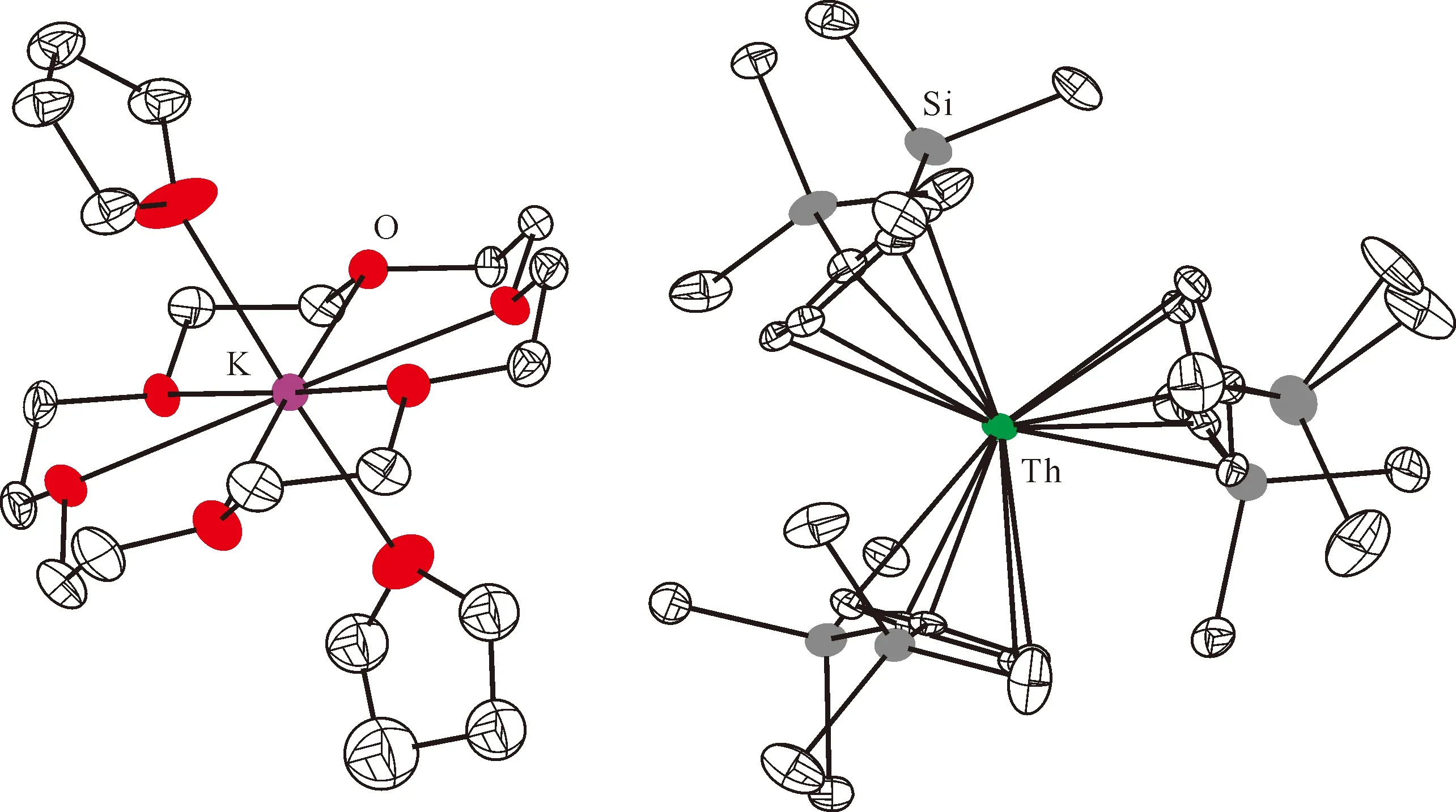
图3 [K(18-c-6)(THF)2][Cp″3Th]的单晶结构(35%热椭球图,氢原子已省略)[60]Fig.3 Thermal-ellipsoid representation of [K(18-c-6)(THF)2][Cp″3Th] (35% probability, hydrogen atoms omitted) [60]
2.2 U(Ⅱ)
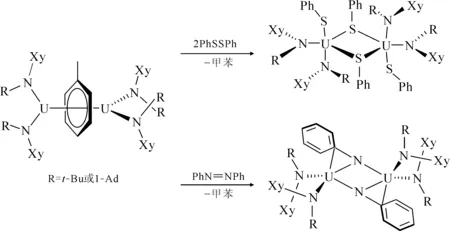
图4 [U(N[R]Xy)2]2(μ-η6,η6-C7H8)(R=t-Bu、1-Ad)与过硫化物或偶氮苯的反应[9]Fig.4 Reactivity of [U(N[R]Xy)2]2(μ-η6,η6-C7H8)(R=t-Bu, 1-Ad) toward PhSSPh and PhNNPh[9]
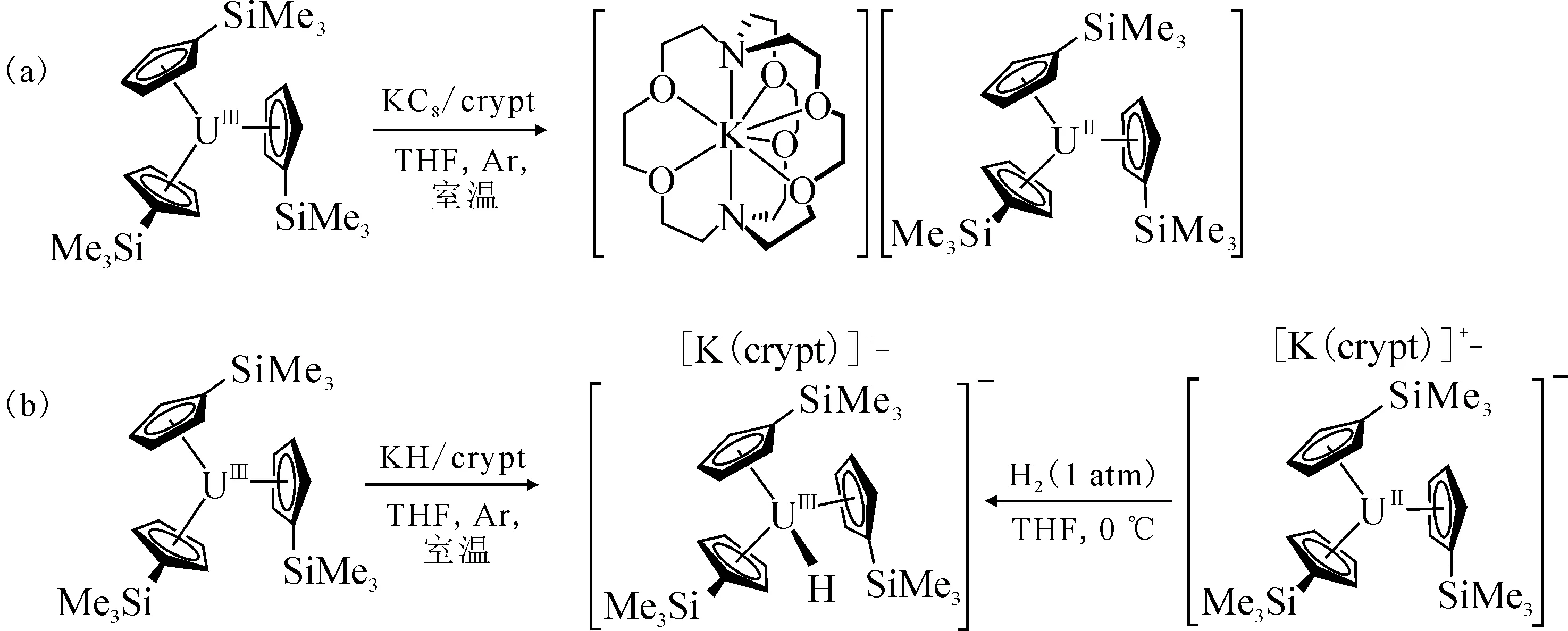
图5 [K(crypt)][Cp′3U] 的合成(a)和 [K(crypt)][Cp′3UH](b)的两种合成路径[106]Fig.5 Synthesis of [K(crypt)][Cp′3U](a) and two synthetic routes for [K(crypt)][Cp′3UH](b) [106]
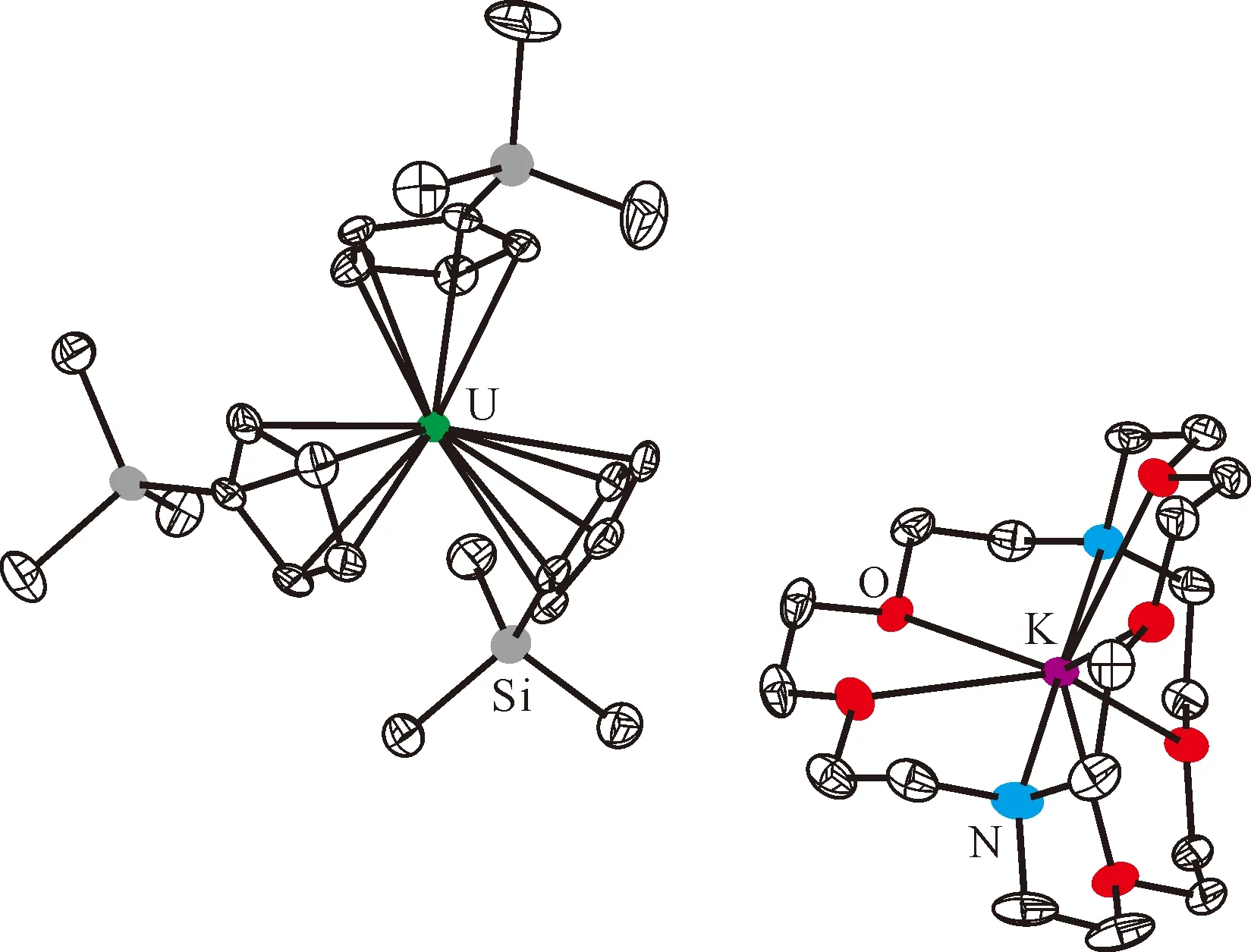
图6 [K(crypt)][Cp′3U]的单晶结构(50%热椭球图,氢原子已省略)[106]Fig.6 Thermal-ellipsoid representation of [K(crypt)][Cp′3U] (50% probability, hydrogen atoms omitted)[106]
2014年,La Pierre等[110]利用含有芳烃底座的三(酚氧基)配体制备了C3对称性的U(Ⅲ)配合物[(Ad,MeArO)3mes]U。循环伏安法表征发现,它在-2.495 V(vs. Fc+/Fc)有接近可逆的还原峰,暗示这一配体体系有可能稳定U(Ⅱ)中心。但是,使用KC8或钠镜在室温下处理[(Ad,MeArO)3mes]U的苯溶液后发现,还原发生在芳烃底座上,配体结构中的苄位氢转移后形成了U(Ⅳ)氢化物。用冠醚(18-c-6或15-c-5即15-冠醚-5)处理U(Ⅳ)氢化物,则使氢负离子从铀上转移到芳烃底座的碳上,形成另一种U(Ⅳ)配合物(图7(a))[110]。不过,在低温和穴醚存在的条件下,用钾球处理[(Ad,MeArO)3mes]U的THF溶液则得到了U(Ⅱ)的配合物。得到的暗红色晶体经X射线晶体学证实为U(Ⅱ)配合物,[K(crypt)][((Ad,MeArO)3mes)U](图7(b))。理论计算表明,配合物中U(Ⅱ)的电子构型为5f4,其5f电子与芳烃底座π*轨道之间存在δ反馈作用[10]。这种三脚架配体支撑的U(Ⅱ)配合物的制备,启发了同系列(非传统)低价稀土配合物的合成与研究[111]。
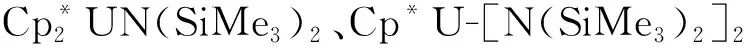
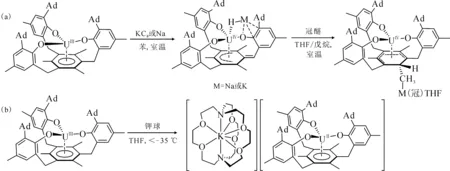
图7 室温下[(Ad,MeArO)3mes]U与还原剂的反应及产物转化 (a)[110]、低温下[(Ad,MeArO)3mes]U与K/crypt反应得到U(Ⅱ)的配合物[K(crypt)][((Ad,MeArO)3mes)U] (b)[10]Fig.7 Decomposition of [(Ad,MeArO)3mes]U under reducing conditions at room temperature(a)[110] and reduction of [(Ad,MeArO)3mes]U to afford U(Ⅱ) complex [K(crypt)][((Ad,MeArO)3mes)U] at low temperature(b)[10]
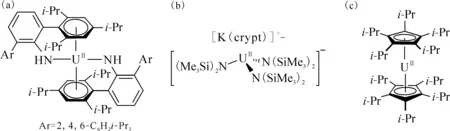
图8 代表性的U(Ⅱ)配合物:U(NHAri-Pr6)2[112](a)、[K(crypt)][U(N(SiMe3)2)3][113](b)和Fig.8 Representative U(Ⅱ) complexes: U(NHAri-Pr6)2[112] (a),
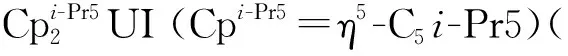
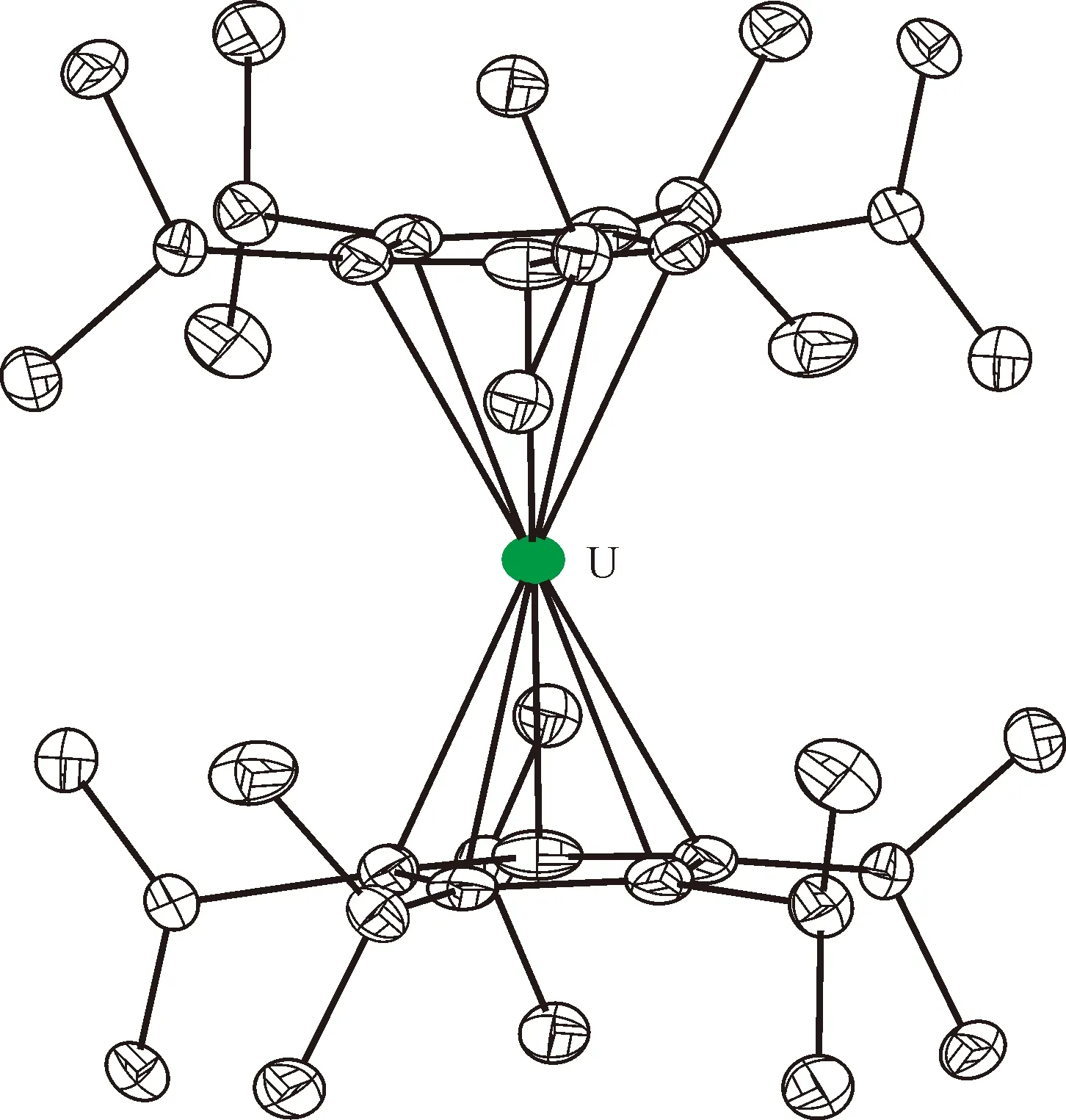
图的单晶结构(35%热椭球图,氢原子和无序碳已省略)[115]Fig.9 Thermal-ellipsoid representation of (35% probability, hydrogen atoms and disordered parts omitted)[115]
近年来,我国学者在低价铀配合物的理论计算与预测方面有较多贡献。潘清江、石伟群课题组通过DFT计算预测了一些配体体系下双核二价铀(UⅡ)2的可能电子构型,包括低聚吡咯大环负四价阴离子、八齿氮基配体等。计算结果表明,(UⅡ)2具有三重态基态,其电子组态为 π4σ2δ2,而铀的5f轨道对高占据分子轨道有重要贡献。在配体支撑的(UⅡ)2分子中铀-铀键的键长为2.33 Å,其Mayer键级为3.89,据此被指认为U(Ⅱ)-U(Ⅱ)的四重键[117]。此外,通过对不同苯环-吡咯环(或吡啶环)组成的混合大环负价配体与铀形成的配合物的U(Ⅲ)/U(Ⅱ)电对的研究,潘清江课题组发现,基于三个苯环与一个吡咯环的负一价配体支撑的U(Ⅱ)配合物,最有希望在合成上实现。其中,铀和芳烃之间的δ键是稳定U(Ⅱ)中心的重要因素[118]。利用几种经典配体体系来稳定U(Ⅰ)配合物的可能性也得到了分析。计算结果表明,Sessler等[119]发展并被 Arnold课题组应用于镎化学的二苯二吡咯大环配体(见2.3节)[120]与U(Ⅰ)形成的配合物在能量上最有利,其中U(Ⅰ)的基态价电子构型为5f5[121]。
2.3 Np(Ⅱ)
近年来,Np(Ⅱ)的金属有机化学取得了一系列进展。2016年,Dutkiewicz等[120]在DME中,用NaK3还原一种基于苯环和吡咯环骨架的大环配位的Np(Ⅲ)氯化物时,得到一种介稳的黑色溶液。Vis-NIR吸收光谱显示,在600 nm和1 275 nm处有新的d→f、d→π*的特征跃迁峰,暗示着Np(Ⅱ)配合物的生成。但是,该体系中无法得到适合表征的单晶产物。放置一段时间后,溶液颜色逐渐转变为棕红色,并且出现了Np(Ⅲ)的谱学特征[120]。
不久后,Dutkiewicz等[122]发现,Cp′3Np和crypt的THF/乙醚溶液在通过石墨钾柱后,颜色由棕绿色变为暗黑色。在低温(195 K)下,溶液中析出的黑色小晶体很可能是Np(Ⅱ)的[K(crypt)]-[Cp′3Np]。但是,由于这种黑色产物在晶态和溶液相中均很不稳定,对它的后续表征与分析难以完成[122]。
2018年,Su等[123]利用金属镎与碘单质在乙醚中反应得到棕色的NpI3(Et2O)x,随后将其与KCp″反应(摩尔比为1∶3)得到了棕绿色的Cp″3Np。在crypt存在的条件下,用KC8还原Cp″3Np(n(KC8)∶n(Cp″3Np)=1.1∶1),得到了首例单晶结构表征的Np(Ⅱ)配合物[K(crypt)]-[Cp″3Np](图10)。由于Np(Ⅱ)与氟具有很强的亲和性,反应过程中需要避免使用聚四氟乙烯磁子。此外,为防止在过滤过程中Np(Ⅱ)被重新氧化,分离时用到的滴管柱中需要负载过量的KC8。这些实验细节凸显了Np(Ⅱ)配合物在制备上的难度。理论计算研究表明,配合物中Np(Ⅱ)的基态电子构型为5f46d1,而非传统认为的5f56d0[123]。
2.4 Pu(Ⅱ)
用KC8处理含有crypt的Cp″3Pu乙醚溶液后,溶液由蓝色变为暗紫色。经处理后可以得到深色的块状晶体,SXRD证实其组成为[K(crypt)]-[Cp″3Pu](图11[124])。这是首例结构表征的Pu(Ⅱ)分子配合物,其核磁氢谱中的化学位移明显受到顺磁中心的影响。由于极易被氧化分解,Pu(Ⅱ)配合物在核磁管内稳定的时间十分有限,不足以完成29Si NMR谱的测量。DFT计算表明,[Cp″3Pu]-离子中Pu(Ⅱ)的基态电子构型以5f66d0为主,混杂了少许5f56d1的贡献[124]。
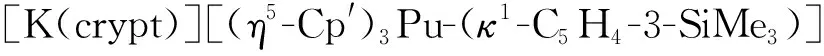
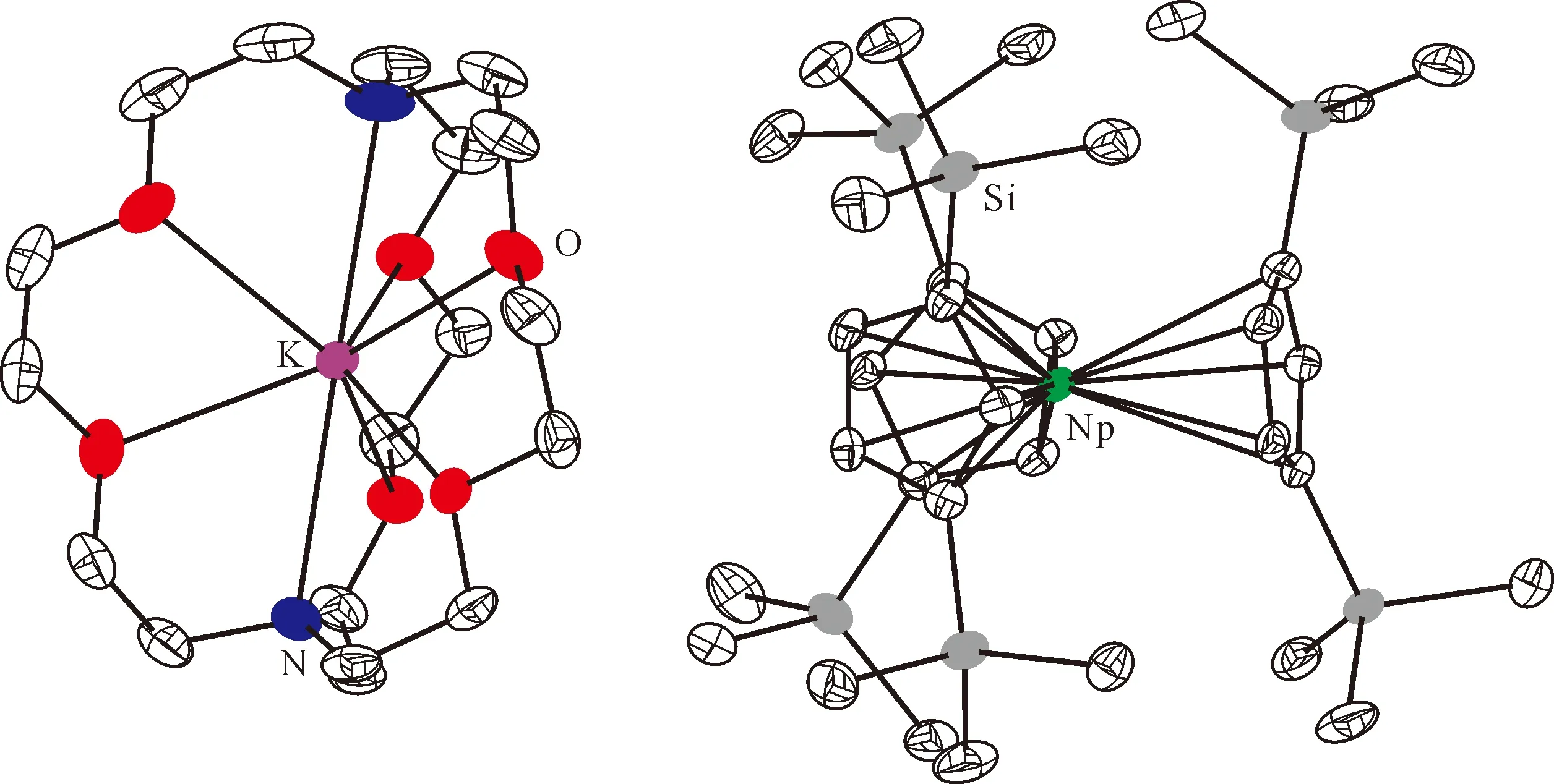
图10 [K(crypt)][Cp″3Np]的单晶结构(50%热椭球图,氢原子已省略)[123]Fig.10 Thermal-ellipsoid representation of [K(crypt)][Cp″3Np] (50% probability, hydrogen atoms omitted)[123]
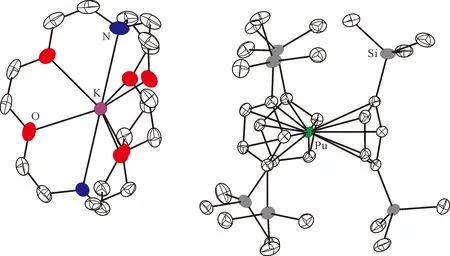
图11 [K(crypt)][Cp″3Pu]的单晶结构(50%热椭球图,氢原子已省略)[124]Fig.11 Thermal-ellipsoid representation of [K(crypt)][Cp″3Pu] (50% probability, hydrogen atoms omitted)[124]
2.5 其他前锕系元素
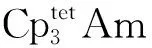

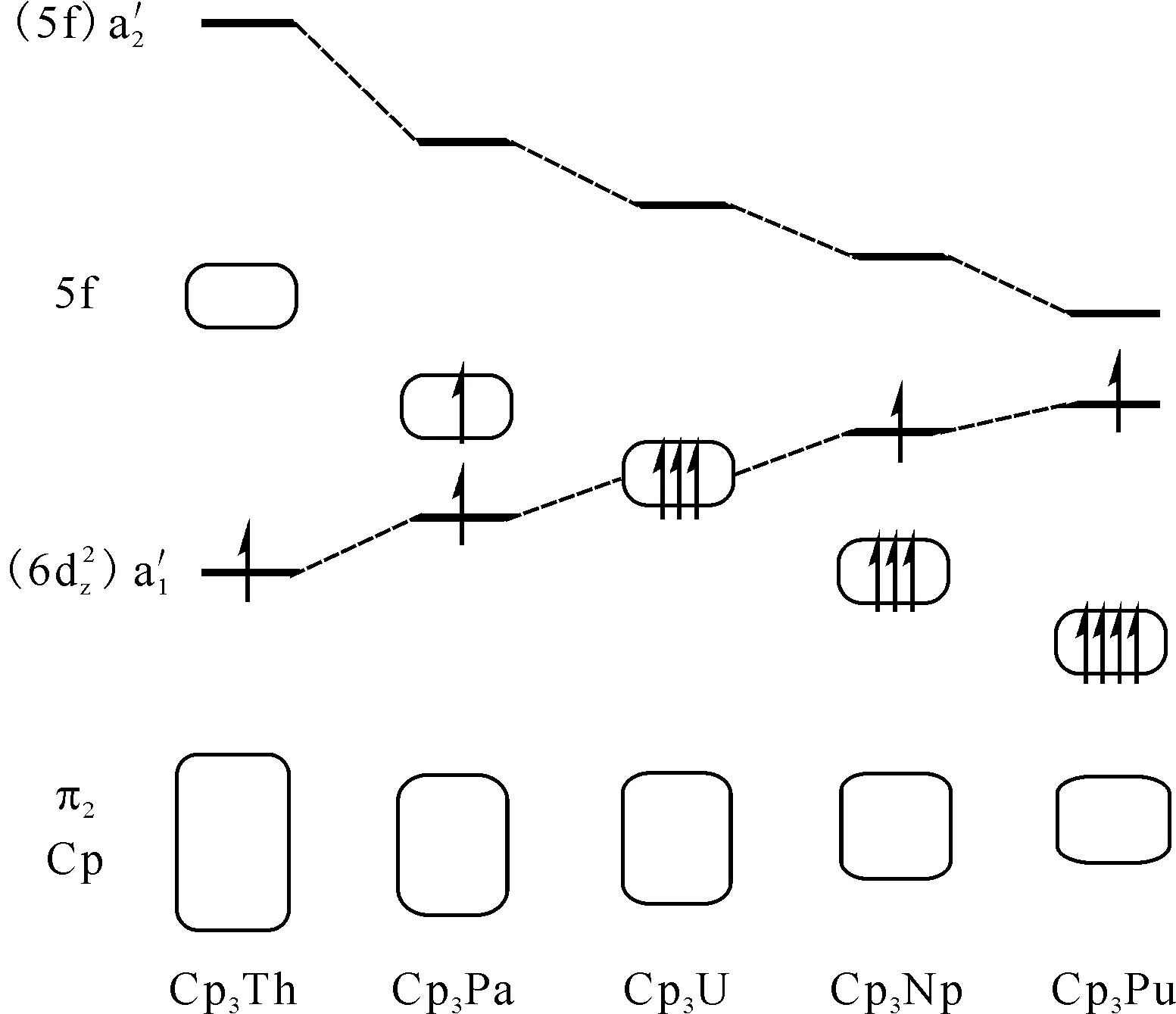
图12 近似D3h构型的Cp3M(M=Th、Pa、U、Np、Pu)的价轨道能级图[51]Fig.12 Energy diagram for valence orbitals of Cp3M (M=Th, Pa, U, Np, Pu) in pseudo-D3h symmetry[51]
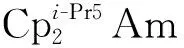
此外,潘清江课题组还利用相对论DFT研究了吡咯环-苯环组成的大环配体(L2-)支撑的二价前锕系元素(不含镅)的结构及其氧化还原性质[144]。结果表明,(MⅢL)+/(ML)电对的还原电势,随着原子序数增加从锕的-2.45 V(vs. Fc+/Fc)升高到钚的-1.64 V(vs. Fc+/Fc)。根据态密度图,对于铀前元素(锕、钍、镤)的中性配合物而言,HOMO主要体现为配体苯环部分的轨道特征,其来自金属的贡献很小。对于这些中性配合物(ML)(M=Ac、Th、Pa)的电子构型的准确描述应为(M3+L3-)。随着原子序数的增加,5f电子在铀、镎、钚的中性配合物(UL、NpL、PuL)中表现出愈发明显的类核性质,而HOMO也逐渐变为金属轨道为主。因此,此类中性配合物(ML)(M=U、Np、Pu)可以被指认为M(Ⅱ)的配合物[144]。
2.6 后锕系元素
目前有单晶结构表征的锔的分子配合物仅有八配位或九配位的Cm(Ⅲ)配合物[128, 145-150]。而锫则仅有两例Bk(Ⅲ)的均配物得到结构表征[131, 151]。2010年,Apostolidis等[152]以1 mg249Cf2O3为起始物,得到了[Cf(H2O)9][CF3SO3]3的单晶。近年来,还有一些锎的分子配合物被报道[131, 147-148, 153]。锕系的最后五个人造元素(锿、镄、钔、锘、铹)目前尚无分子配合物的单晶结构报道。考虑到制备反应的低核截面产率与产生的核素的短寿命,许多超锎核素的实验依赖于一些特殊技术和方法(如单次单原子技术)[154-156],这无疑限制了超锎元素分子配合物的研究。因此,对于这些后锕系元素的非寻常氧化态的分子配合物而言,DFT计算是目前占主导地位的研究方法[157]。
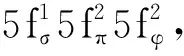
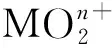
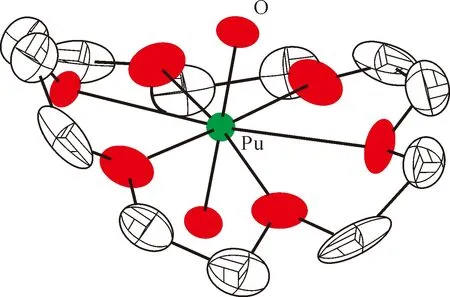
图13 [PuO2(18-c-6)][ClO4]的单晶结构(35%热椭球图,氢原子和抗衡阴离子[ClO4]-已省略)[163]Fig.13 Thermal-ellipsoid representation of [PuO2(18-c-6)][ClO4] (35% probability, hydrogen atoms and counter anion [ClO4]- omitted)[163]
尽管受限于实验条件,非寻常氧化态的后锕系元素分子配合物仍有一些实例。比如,水溶液中合成的四价锫配合物Bk[3,4,3-LI(1,2-HOPO)]就是一个例子[170]。此外, Albrecht-Schmitt课题组于2015年利用249CfCl3与过量2,6-吡啶二甲酸(H2DPA)反应得到了Cf(HDPA)3·H2O的晶体。针对这一配合物的磁学、光谱学等研究表明,Cf(Ⅲ)配合物的光致发光机理并非源自5f-5f跃迁,而是配体和金属之间的电荷转移跃迁。具体地说,Cf(Ⅲ)通过LMCT被还原为Cf(Ⅱ),同时在价带产生一个正电空穴(h+)。当介稳态的Cf(Ⅱ)与h+再复合回到Cf(Ⅲ)的基态时,便有光子放出。对于那些配位体系中难以形成h+的化合物,比如K[Cf2(SO4)2F3],则未观察到光致发光现象,这进一步验证了该机理的合理性[147]。刘国奎等[171]通过分析指出,较强的离子-配体相互作用使得电荷转移跃迁态的能级降低至接近价带水平,因而促进了Cf(Ⅱ)的形成。
3 总结与展望
随着非水溶剂配位化学的发展[1],化学家们已经能够将+2氧化态扩展至钍[60]、铀[106]、镎[123]、钚[124]的分子配合物,并通过SXRD获得可靠的结构数据。引人瞩目的是,锕系元素的非寻常氧化态和稀土金属的非传统氧化态化学有了更多的联动性,二者互为参考、互相促进。比如,受茂基配体稳定非传统二价稀土金属的启发,Evans[172]随后报道了其在支撑二价锕系金属配合物中的应用。另一方面,Meyer课题组首先发展了以芳烃为底座的三脚架型酚氧配体在锕系金属配位化学中的应用[10, 173],随后通过与Evans等合作将其拓展到了稀土金属的配位化学中[111, 174-176]。
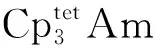
综上,锕系元素的非寻常氧化态研究无疑是当前锕系元素配位化学与金属有机化学中,最富生机、最具潜力、同时也最具挑战的领域之一。合成化学在这一领域的发展中起到了关键性的作用,但仍需其他方向上的协作才能取得更大进展。X射线吸收精细结构(EXAFS)、X射线近边结构(XANES)、磁圆二色谱(MCD)、EPR、SQUID等表征手段将在探究锕系元素非寻常氧化态的电子结构与磁学性质方面发挥越来越重要的作用。未来,锕系元素非寻常氧化态的研究将不仅限于基础研究,也将在诸多应用领域,如核燃料后处理、单分子磁体等方面展现巨大潜力[178]。通过更广泛、更深入的交流与合作,我们期待并坚信中国学者和中国团队会在锕系元素的非寻常氧化态化学和相关学科交叉领域做出更多的贡献。
猜你喜欢
无线电工程(2022年10期)2022-10-24
数学物理学报(2022年5期)2022-10-09
中国特种设备安全(2022年4期)2022-07-08
人工晶体学报(2022年3期)2022-04-14
粉末冶金技术(2021年3期)2021-07-28
延边大学学报(自然科学版)(2021年1期)2021-04-27
量子电子学报(2021年2期)2021-04-24
人工晶体学报(2019年5期)2019-06-18
当代陕西(2019年6期)2019-04-17
分析化学(2018年2期)2018-03-02
