
UPLC-MS/MS同时测定不同产地石仙桃中30种酚类成分的含量
2021-03-02 09:26林逸凡张宗伟刘剑任铭诚许文赵海誉黄鸣清
药学研究 2021年1期
林逸凡,张宗伟,刘剑*,任铭诚,许文,赵海誉,黄鸣清*
(1.福建中医药大学药学院,福建 福州 350122;2.中国中医科学院中药研究所,北京 100700)
石仙桃(PholidotachinensisLindl.)为兰科石仙桃属植物石仙桃的假鳞茎或全草,始载于《生草药性备要》[1],主要分布于福建、广东、云南等地。石仙桃具有养阴,清肺,利湿,消瘀的功效,民间常用于治疗头痛、咳嗽、高血压等,其单方制剂头痛定糖浆临床上用于治疗神经功能性头痛、脑震荡后遗症[2]。化学研究表明[3-6],石仙桃中含有黄酮类、菲类、萜类、多糖等组分,但这些研究目前主要集中在多糖、菲类化合物的分离鉴定,很少涉及以黄酮为代表的酚类成分。而大量研究表明[7-14],石仙桃中黄酮等酚类成分具有局麻、镇痛、抗氧化、抗惊厥、抗肿瘤、抗疲劳、抗缺氧等多种活性,提示酚类成分可能是其主要药效物质之一。
在质量评价方面,刘建新等[15]采用紫外可见分光法测定石仙桃总黄酮的含量;张淼等[16]采用高效液相色谱(HPLC)法对不同产地的石仙桃药材进行指纹图谱研究;林丽聪等[17]采用HPLC法对石仙桃中的两个酚类成分(天麻素和天麻苷元)进行含量测定;侯慧静等[18]采用超高效液相色谱(UPLC)法测定石仙桃中两个二氢菲类化合物(2,7-二羟基-3,4,6-三甲氧基-9,10-双氢菲和coelonin)的含量。然而,上述研究仅围绕总黄酮或个别酚类化合物,并未整体考虑石仙桃中各种酚类成分。此外,石仙桃分布广泛,在各地区以野生为主,药材质量参差不齐,有必要对其进行多成分质量控制。因此,本研究建立了一种超高效液相色谱串联质谱(UPLC-MS/MS)同时测定石仙桃中30种酚类成分含量的方法,并对不同产地的石仙桃药材进行定性定量及聚类分析,将为深入阐明石仙桃药效物质及质量控制提供实验依据。
图1 各成分及内标物化学结构图
1 材料
Acquity UPLC I-Class超高效液相色谱仪(美国Waters公司);Xevo TQ-S三重四极杆质谱(美国Waters公司);Milli-Q超纯水仪(美国Millipore公司);CPA225D型十万分之一分析天平(德国Sartorius公司);JXFSTPRP-24研磨仪(上海净信科技有限公司);Heraeus Fresco17离心机(美国Thermo Fisher Scientific公司);HH-1恒温水浴锅(国华电器有限公司);KQ-500E台式超声波清洗器(昆山市超声仪器有限公司)。
乙腈、甲醇、水为质谱纯(德国Merck公司);甲酸为色谱纯(上海阿拉丁生化科技股份有限公司),其余试剂均为分析纯;对照品香草酸(批号:MUST-16062307)、阿魏酸(批号:MUST-16021902)、丁香酸(批号:MUST-17041910)、儿茶素(批号:MUST-16081503)、鞣花酸(批号:MUST-16062603)、葛根素(批号:MUST-15012916)、荭草苷(批号:MUST-13070502)、异荭草苷(批号:MUST-13100902)、牡荆素-2-O-鼠李糖苷(批号:MUST-17101202)、原儿茶醛(批号:MUST-15040502)、咖啡酸(批号:MUST-17032010)、白杨素(批号:MUST-14070310)、根皮素(批号:MUST-15012608)、木犀草素(批号:MUST-17102605)、木犀草苷(批号:MUSE-17030405)、金丝桃苷(批号:MUST-17022010)、洋蓟素(批号:MUST-16041015)、夏佛托苷(批号:MUST-14092002)、异夏佛托苷(批号:MUST-14060203)、水仙苷(批号:MUST-17040109)、反式-2-羟基肉桂酸(批号:MUST-16051803)、野黄芩苷(批号:MUST-13091204)均购于成都曼思特生物科技有限公司;牡荆素(批号:111687-200602)、槲皮苷(批号:111538-200504)、芦丁(批号:100080-201610)、绿原酸(批号:110753-201716)购于中国药品生物制品检定所;紫云英苷(批号:13022721)、异绿原酸C(批号:13022523)、山柰酚-3-O-芸香糖苷(批号:13103022)购于天津仕兰科技有限公司;芹菜素(批号:Y131214)、没食子酸乙酯(批号:Y103947)购于北京赛佰草科技有限公司;紫铆素(批号:T09J7Z17539)购于上海源叶生物科技有限公司;以上对照品均为HPLC级,含量 ≥ 98.00%。
13批石仙桃药材分别采集于浙江丽水(S1)、福建宁德(S2)、福建泉州(S3)、四川成都(S4)、广东韶关(S5、S9、S13)、福建漳州(S6、S7)、广西玉林(S8)、广西钦州(S10)、广东揭阳(S11)、云南曲靖(S12),并经福建中医药大学黄泽豪教授鉴定为兰科石仙桃属植物石仙桃(PholidotachinensisLindl.)的假鳞茎或全草。
2 方法与结果
2.1 色谱条件 色谱柱:Welch Xtimate UHPLC C18(2.1 mm×100 mm,1.8 μm);流动相:0.1%甲酸水(A)-乙腈(B),采用梯度洗脱模式,流动相比例为:0~0.5 min,2% B;0.5~2.5 min,2%→7% B;2.5~4.5 min,7%→13% B;4.5~6.5 min,13%→20% B;6.5~9.5 min,20%→55% B;9.5~12.5 min,55%→90% B;12.5~13.5 min,90% B;13.5~13.6 min,90%→2% B;13.6~15 min,2%→2% B;流速:0.2 mL·min-1;柱温:45 ℃;进样量:2 μL。
质谱条件:采用正负切换的多反应监测(MRM)模式,离子源为电喷雾电离源,质谱参数如下:离子源温度150 ℃,脱溶剂气流量800 L·h-1,脱溶剂气温度500 ℃,毛细管电压2.5 kV,脱溶剂气N2,碰撞气体Ar,锥孔气流量50 L·h-1,驻留时间0.02 s。待测成分及内标物的质谱优化参数见表1,各成分MRM色谱图见图2。
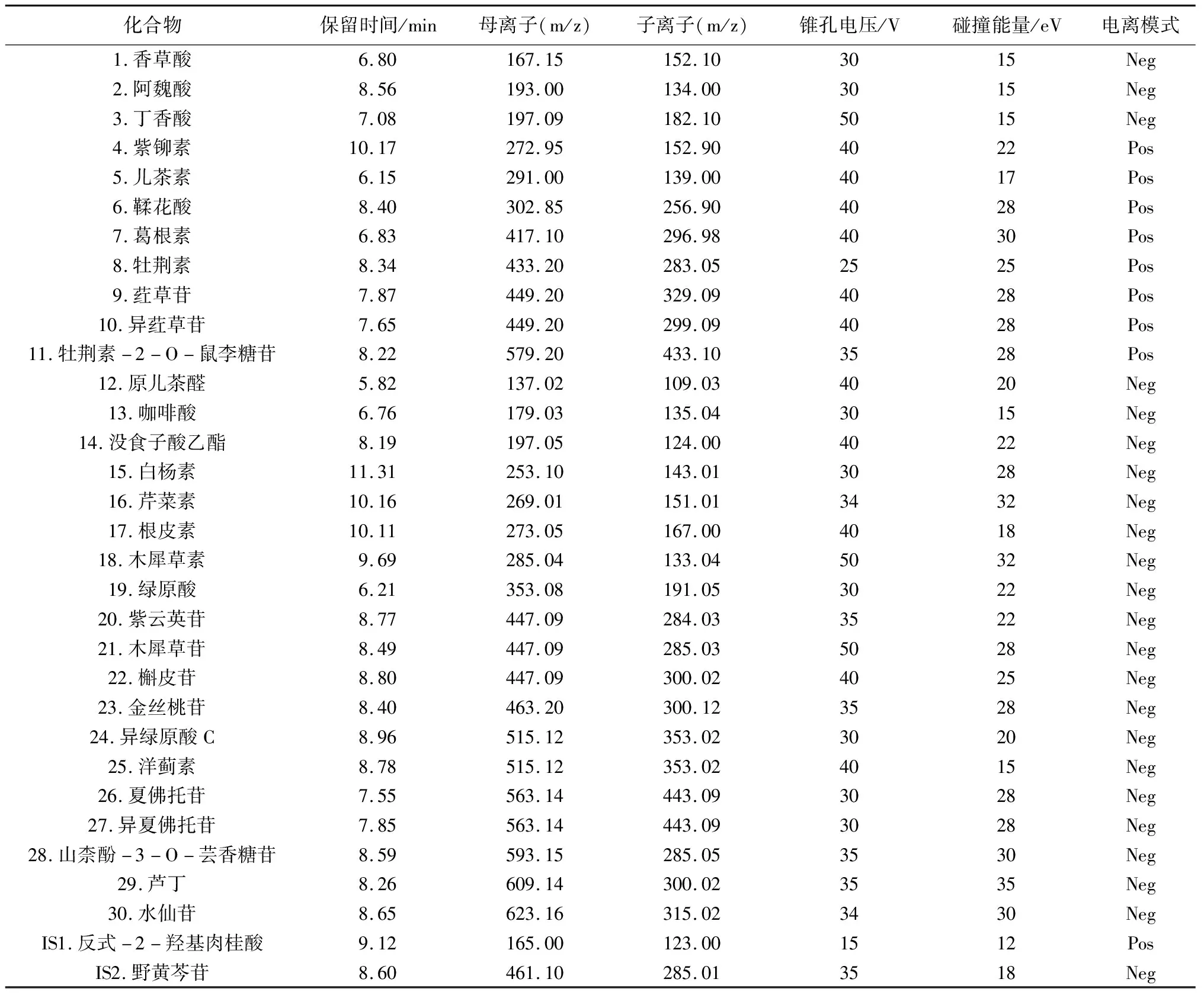
表1 各化合物及内标物质谱优化参数

图2 各成分及内标物的MRM色谱图
2.2 溶液的制备
2.2.1 混合对照品溶液的制备 分别精密称定香草酸、阿魏酸、丁香酸、紫铆素、儿茶素、鞣花酸、葛根素、牡荆素、荭草苷、异荭草苷、牡荆素-2-O-鼠李糖苷、原儿茶醛、咖啡酸、没食子酸乙酯、白杨素、芹菜素、根皮素、木犀草素、绿原酸、槲皮苷、紫云英苷、木犀草苷、金丝桃苷、异绿原酸C、洋蓟素、夏佛托苷、异夏佛托苷、山柰酚-3-O-芸香糖苷、芦丁、水仙苷对照品适量,加入纯甲醇溶解并稀释至刻度,制备为浓度2 mg·mL-1的单一对照品储备液。分别精密量取各对照品储备液适量,用50%甲醇溶液稀释至刻度,摇匀,分别得到质量浓度为4 014.78、4 073.7、2 433.2、504、19.76、210、102、4 075.5、2 056.824、2 005.248、10.022 4、1 054.85、509.76、120.4、203.84、2 068、10.128、698.28、200、62.01、408、84、1 833.96、424.2、304.92、247.68、524、1 666.98、783、2 021.376 ng·mL-1的混合对照品溶液。
2.2.2 内标溶液的制备 精密称定对照品反式-2-羟基肉桂酸、野黄芩苷适量,加入纯甲醇溶解并稀释至刻度,制备为浓度2 mg·mL-1的单一对照品储备液,分别精密量取内标对照品储备液适量,用50%甲醇溶液稀释至刻度,摇匀,得到浓度分别为367.8、836.8 ng·mL-1的内标混合溶液。
2.2.3 供试品溶液的制备 石仙桃全草于60 ℃烤箱中干燥48 h后,粉碎,过筛。精密称取1 g粉末,置于锥形瓶中,加入40 mL 80%乙醇,摇匀,于80 ℃水浴锅中回流提取2 h,静置冷却,离心(10 000 r·min-1)10 min,取上清液,经0.22 μm微孔滤膜滤过,取续滤液即得。用于定量分析的样品溶液:取续滤液稀释10倍,按1∶1(200∶200,μL)加入内标溶液后进行测定。
2.3 方法学考察
2.3.1 线性关系考察及定量限测定 精密量取“2.2.1”项下的混合对照品溶液,用50%甲醇逐级稀释,并加入内标液,即得一系列不同浓度的标准溶液。以各成分与内标峰面积比值(Y)为纵坐标,待测成分浓度(X)为横坐标,进行线性回归。分别以信噪比为3和10时各对照品溶液的浓度作为检测限(LOD)和定量限(LOQ),见表2。
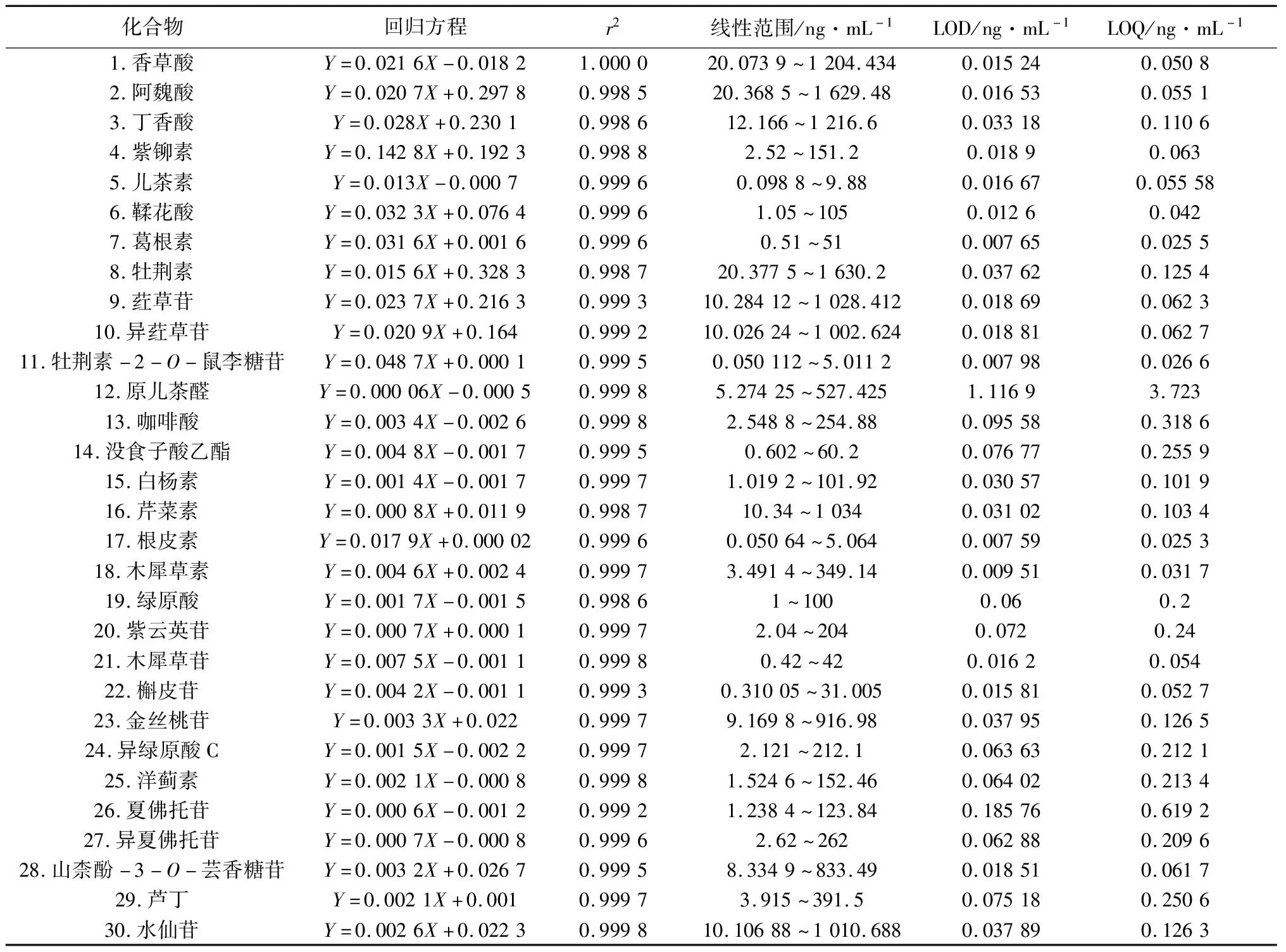
表2 各检测成分的回归方程、相关系数、线性范围、检测限和定量限
2.3.2 精密度试验 配制适当浓度的混合对照品溶液,在“2.1”和“2.2”项下的色谱与质谱条件下,当天连续进样6次测定其日内精密度,连续测定3 d,考察其日间精密度,记录各对照品与内标的峰面积比,并计算RSD,结果见表3,RSD均小于5.19%,表明仪器精密度良好。
2.3.3 稳定性试验 取石仙桃药材适量,按“2.2.3”项下制备供试品溶液,分别于制备后的0、2、4、8、12、24 h进样测定其峰面积,计算RSD,结果见表3,RSD均小于3.80%,表明供试品溶液在24 h内稳定。
2.3.4 重复性试验 取同一批石仙桃药材,按“2.2.3”项下同法制备供试品溶液6份,分别进样测定,计算其平均含量及RSD,结果见表3,RSD均小于4.12%,表明方法重复性良好。
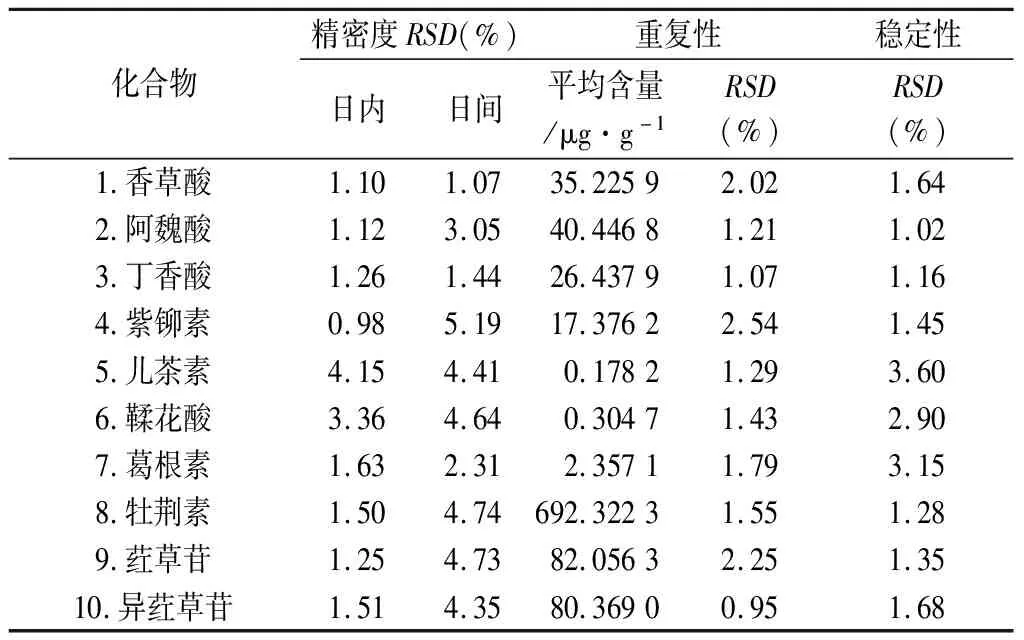
表3 精密度、重复性、稳定性试验结果
2.3.5 加样回收率试验 精密称定已知含量的石仙桃药材适量,平行9份,分别精密加入低、中、高3种浓度的对照品溶液,按“2.2.3”项同法制备供试品溶液,分别进样测定,计算平均回收率及RSD,结果见表4。
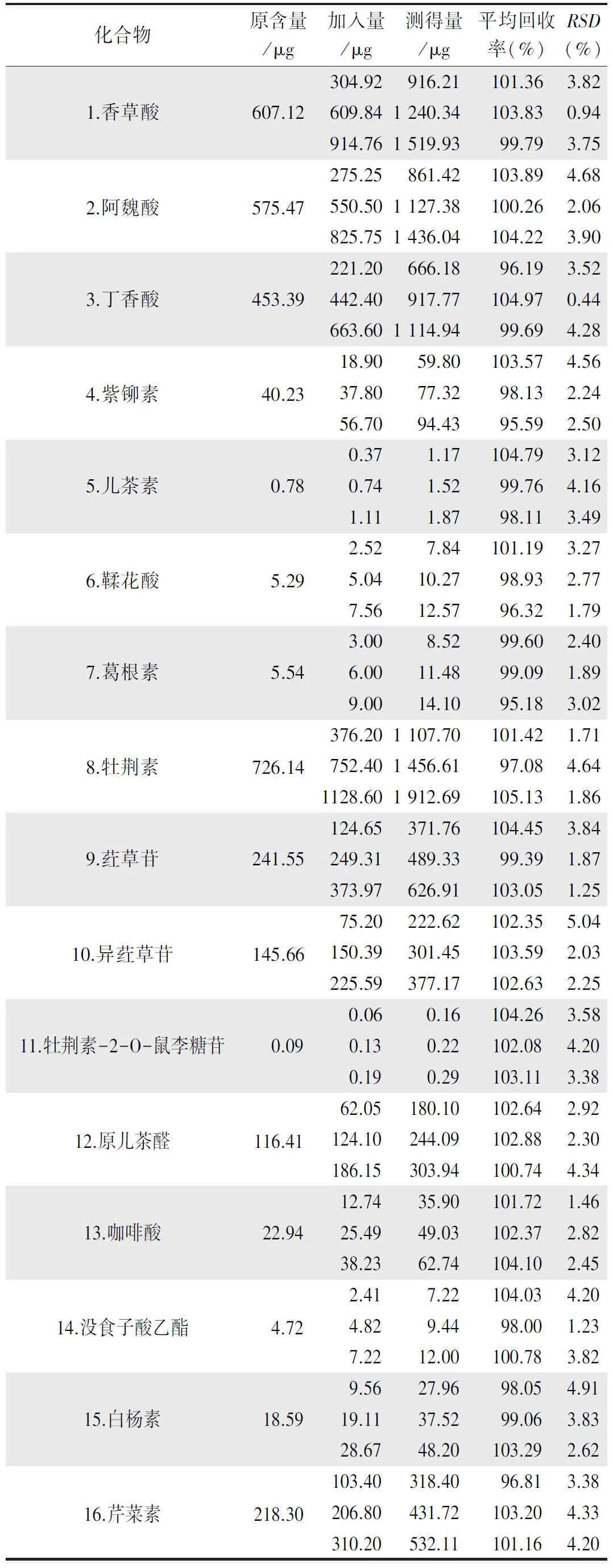
表4 各成分回收率考察结果
2.4 样品含量测定 分别精密称取不同产地石仙桃药材适量,按“2.2.3”项下的方法制备样品,再按“2.1”和“2.2”项下的LC-MS条件进样测定,根据内标法计算各批次药材中30种化合物的含量,结果见表5。
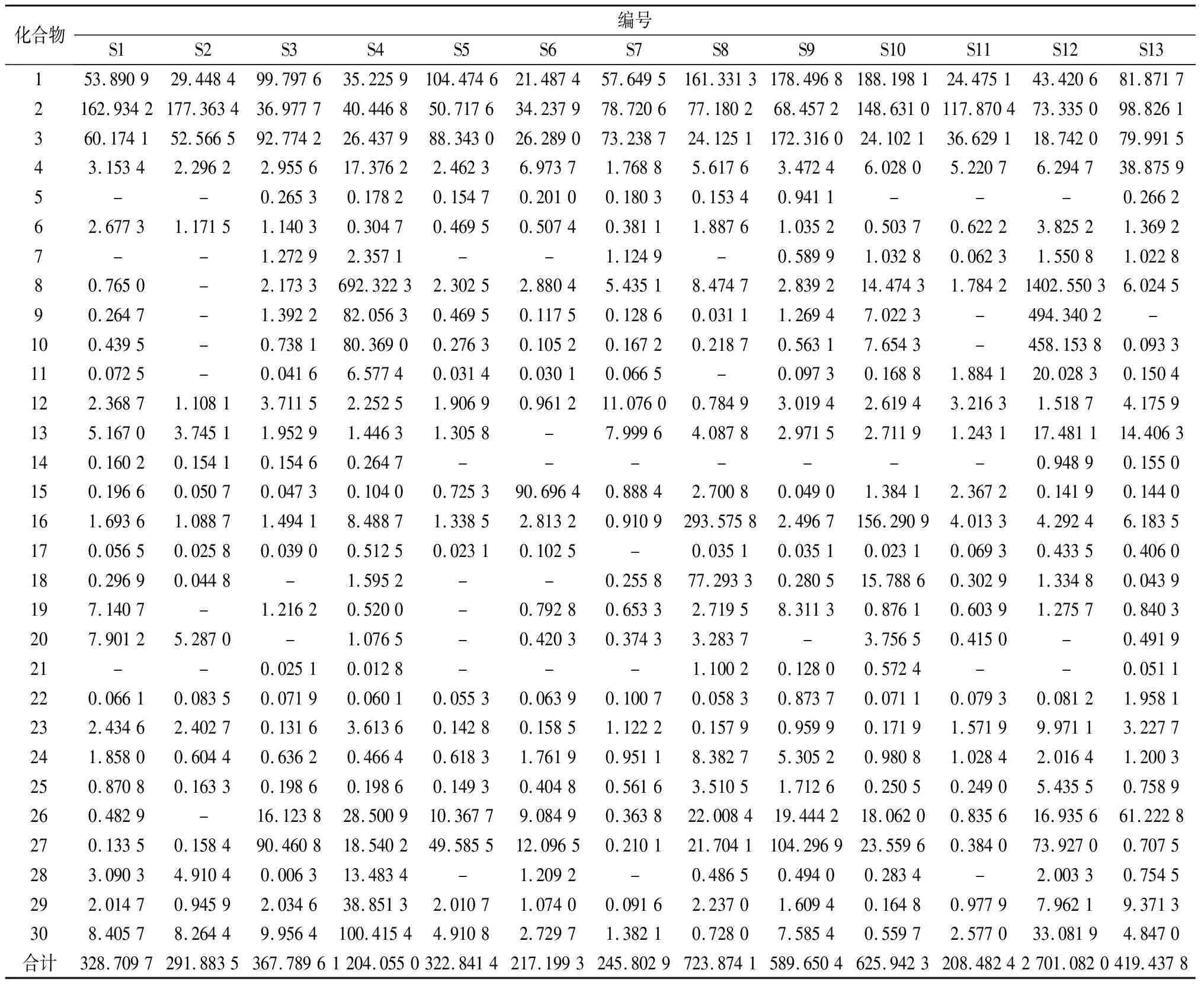
表5 不同产地药材含量测定结果(μg·g-1,n=3)
2.5 聚类分析 为考察不同产地药材间的差异性,采用SPSS 26.0软件,以本法测定的30种化合物含量为变量,以Squared Euclidean Distance为测度,对13批不同产地的石仙桃药材进行聚类分析,结果显示(图3),13批药材可聚为2类,其中浙江(S1)、福建(S2、S3、S6、S7)、广东(S5、S9、S11、S13)、广西(S8、S10)产地归为Ⅰ类,四川(S4)与云南(S12)产地归为Ⅱ类,可见不同地域的石仙桃药材在质量方面存在一定的差异性。
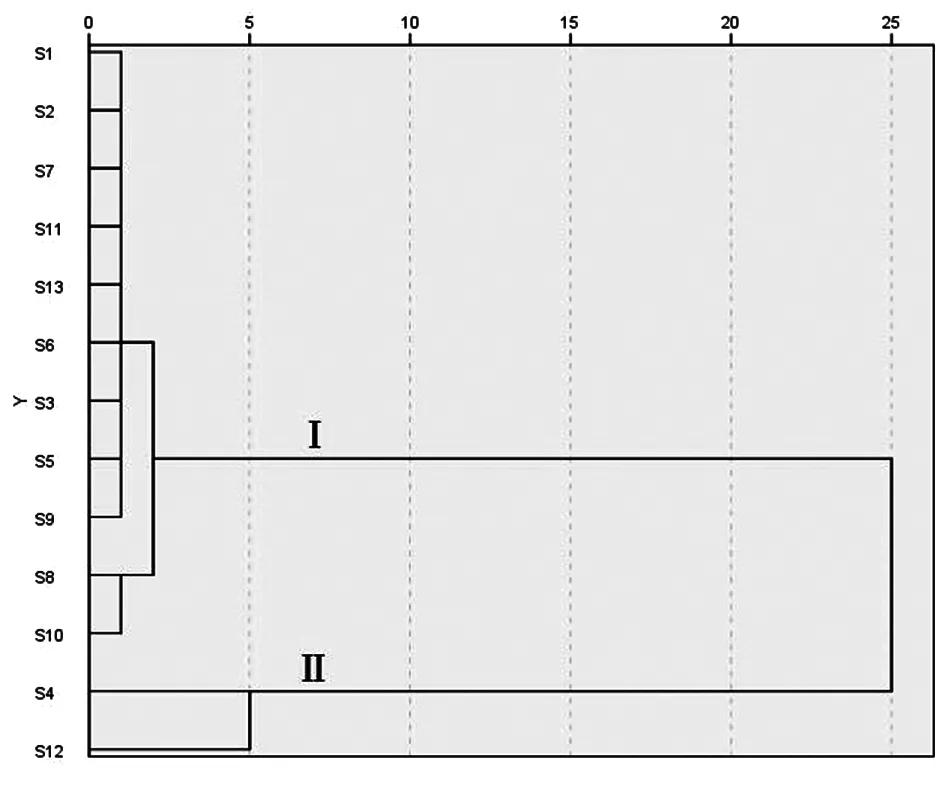
图3 13批不同产地石仙桃药材的层次聚类图
3 讨论
3.1 色谱条件的优化 为快速、有效分离各成分,前期比较了不同规格色谱柱的分离效果。结果显示,Welch Xtimate UHPLC C18柱(2.1 mm×100 mm,1.8 μm)对大部分化合物的分离效率较高,且峰型尖锐,对称性好,适用于本试验成分体系的测定。另一方面,通过比较甲醇-水、乙腈-0.1%甲酸水溶液等4种流动相体系的洗脱效果,发现在含甲醇的流动相体系下色谱峰存在严重的拖尾与基线漂移现象,且峰型较宽,洗脱能力远不如含乙腈的流动相体系;而在流动相中加入甲酸,可减少拖尾,改善峰型,促进质子化,增强离子响应,故选用乙腈-0.1%甲酸溶液作为流动相。
3.2 质谱条件的优化 为了使离子响应达到最高,分别采用正、负离子模式对各成分的单一对照品溶液进行检测,发现多数化合物在负离子模式下响应较高,而个别化合物仅在正离子模式下有响应,因此采用正、负离子切换模式,保证各成分在最佳电离模式下进行测定[19]。此外,对各化合物进行二级质谱扫描,在MRM模式下,通过能量碰撞的方式选择响应最高的碎片离子作为定量离子(见表1)。以葛根素为例,其一级离子为417.1,经碰撞裂解,产生399.0、351.2、321.1、296.9多个二级离子,其中响应最高且干扰最小的是296.9,即[C18H16O4+]的碎片离子峰,故选其为葛根素的定量离子。
3.3 不同产地药材含量分析 通过对不同产地药材进行含量测定,发现各批次间的成分含量存在差异;结合聚类分析可知,沿海产地(浙江、福建、广东、广西)药材质量较一致,但与内陆产地(四川、云南)差异较大。进一步分析发现,内陆产地药材中的牡荆素、荭草苷、异荭草苷含量较高,且成分总量显著高于沿海产地药材。由此推测石仙桃中部分酚类成分含量高低可能与产地地理位置、气候土壤等因素相关。综上可见,不同地区的石仙桃药材在质量上存在较大差异,因此有必要对其进行全面的质量控制研究。
3.4 结论 本试验基于UPLC-MS/MS建立了一种同时测定石仙桃多成分的定量分析方法,该方法在15 min内实现了对各成分的基本分离,其精密度、重复性及稳定性良好,具有高选择性、高灵敏度的特点,可用于石仙桃药材的质量评价。此外,聚类分析结果提示不同地区的石仙桃药材质量存在较大差异。
猜你喜欢
航天电子对抗(2022年4期)2022-10-24
中国科技纵横(2020年1期)2020-04-20
中国科技纵横(2019年23期)2019-02-14
学苑创造·B版(2018年2期)2018-01-29
故事作文·高年级(2017年10期)2017-10-19
读者(2017年9期)2017-04-12
海峡科技与产业(2017年1期)2017-03-04
故事作文·高年级(2017年1期)2017-03-01
中国信息化·学术版(2013年3期)2013-06-25
中国医药导报(2011年27期)2011-12-31
