
不同制备方法对氧化铈结构及甲苯催化燃烧性能的影响
2021-02-24 06:15权燕红武朦朦赵金仙
燃料化学学报 2021年2期
权燕红 ,苗 超 ,李 涛 ,王 娜 ,武朦朦 ,张 宁 ,赵金仙 ,任 军
(太原理工大学 煤科学与技术省部共建国家重点实验室培育基地,山西 太原 030024)
挥发性有机物(volatile organic compounds,VOCs)是形成PM2.5 和O3的重要前驱体,近年来成为一种广受关注的重要大气污染物[1]。在诸多VOCs处理技术中,应用较为广泛的主要包括燃烧法、吸附法和冷凝法,其中,燃烧法可将污染物转化为CO2和H2O 最终从环境中消除,具有处理效率高、副产物少、无二次污染等优势,因此,受到学术界和工业界的高度重视。与传统直接热力燃烧技术相比,催化燃烧技术采用催化剂降低反应活化能,起燃温度大幅降低,燃料使用量和设备制造成本减少,具有更广阔的应用前景和市场空间[2]。开发高效稳定的催化剂,使得有机废气尽量在更低的温度下发生燃烧,是催化燃烧技术的关键和核心。
催化燃烧VOCs 主要使用贵金属和过渡金属氧化物催化剂,贵金属(Pt、Pd 和Rh 等)催化剂活性很高,在较低温度下可实现VOCs 的高效分解,寿命较长但价格昂贵[3]。相比之下,开发价格低廉的过渡金属氧化物催化剂受到广泛关注。氧化铈含有丰富的Ce3+/Ce4+离子对以及 Ce3+Ce4+之间的快速可逆转化性能,因而显示出独特的氧化还原特性和极强的储氧能力,显著促进分子氧的吸附和活化[4],因此,作为活性组分或载体可以应用于VOCs 催化燃烧反应[5,6]。
研究者针对不同形貌CeO2的催化燃烧性能展开了大量研究,如Peng 等[7]通过控制水热合成条件得到不同形貌的CeO2(纳米棒、纳米颗粒、纳米立方体),分别负载Pt 后用于催化甲苯燃烧反应,发现以纳米棒CeO2为载体的催化剂表面氧空位浓度最高,催化活性最佳;Feng 等[8,9]研究发现,CeO2空心球比CeO2纳米棒和纳米管在催化甲苯燃烧反应中表现出更高的活性,主要归因于其具有更高的比表面积和更多的氧空位;Liao 等[10]采用水热合成法制备CeO2纳米棒,通过改变水热时间和碱溶液浓度调节样品尺寸,发现直径最小的纳米棒具有最多的氧空位和更高的Ce3+浓度,在甲苯催化燃烧反应中表现出最佳的活性。此外,有研究者认为,CeO2暴露晶面对其催化性能具有重要影响。例如,Lv 等[11]通过控制合成得到具有高度规整的(100)晶面的CeO2立方体,相比暴露(111)和(100)两种晶面的CeO2纳米颗粒,其在甲苯选择性氧化反应中表现出更高的苯甲醛选择性;Huang 等[12]分别通过水热法和沉淀法制备了棒状和颗粒状的CeO2负载Au 用于CO 催化氧化反应,发现棒状CeO2主要暴露(100)和(110)晶面,而颗粒状CeO2主要暴露(111)晶面,由于(100)和(110)晶面比(111)晶面更有利于Au 的锚定与分散,导致棒状CeO2负载Au 催化剂表现出更高的催化活性;Pan 等[13]通过控制水热合成条件制备了不同形貌(棒状、片状和管状)的CeO2,其中,CeO2纳米片兼具二维材料高比表面积、高结晶度及自身优异的储氧和氧化还原特性,表现出最高的CO 催化氧化活性,主要归因于其具有最高的(100)晶面暴露比例;闫宁等[14]采用不同方法制备出球状、花苞状和多面体的CeO2,负载Ni 后用于CO 甲烷化反应,发现球状CeO2比表面积最高,主要暴露(111)晶面,而且氧空位含量高,在反应中表现出优异的催化性能。采用溶胶-凝胶-超临界干燥法可以制得小粒径、高比表面积、低密度的CeO2气凝胶,其丰富的表面缺陷能够提供很高的表面氧浓度。李健[15]采用CeO2气凝胶担载CuO用于富氢气氛中CO 的催化氧化,发现CuO 与CeO2之间存在强烈的相互作用,使铜物种高度分散于CeO2表面上,因此,表现出很高的活性和选择性。
综上分析,氧化铈的催化性能主要取决于比表面积、晶粒尺寸、暴露晶面等关键结构参数,而这些结构性质受到制备方法的影响和控制。研究显示,水热法、沉淀法和溶胶凝胶法在合成高性能CeO2方面具有明显优势。在前期研究基础上选择合适的制备方法,通过优化工艺条件定向合成具有特定形貌的CeO2样品,有望进一步改善催化燃烧性能[16-19]。
本研究采用溶胶-凝胶-超临界干燥法、水热法、沉淀法分别制备不同形貌的氧化铈,探讨了催化燃烧甲苯反应性能,通过多种方法对不同形貌的氧化铈样品的物理和化学结构进行了全面表征,分析了氧化铈样品形貌、暴露晶面等微观结构参数及其与反应物分子的吸附能力和自身的氧化还原循环能力的关系,揭示了其对催化燃烧甲苯性能的影响规律。
1 实验部分
1.1 不同形貌氧化铈的制备
CeO2-A:采用溶胶-凝胶-超临界干燥法合成气凝胶[20]。将硝酸铈溶于30 mL 乙醇中充分溶解后,再依次加入2.63 g 柠檬酸,和10 mL 环氧丙烷,快速搅拌形成均相溶液,于模具中静置形成湿凝胶;得到的湿凝胶在高压反应釜中于进行超临界干燥,临界温度为243 ℃,临界压力为6.4 MPa,密闭,充入高压氮气,检漏,再排出,重复进行充放气以排出反应釜内空气。设定温度为270 ℃进行加热,釜内压力升至平衡(8 MPa),稳定1 h 后,开始缓慢放气直至釜内气体全部排出,停止加热,待反应釜自然冷却降至室温后,取出样品即得干燥的气凝胶。然后以1 ℃/min 的速率升温至300 ℃焙烧,保持2 h,即得氧化铈气凝胶,记为CeO2-A。
CeO2-R:该催化剂由水热法合成。室温剧烈搅拌下,将1.25 g 硝酸铈加入至37.5 mL 甲醛溶液中,使其充分溶解。然后将2.625 g 氢氧化钾缓慢加入至上述溶液中,搅拌均匀;之后,将得到的混合溶液移至不锈钢水热釜中,于100 ℃烘箱中放置20 h,待反应釜自然冷却至室温后,过滤、洗涤,收集样品。经60 ℃空气干燥12 h,450 ℃焙烧2 h,制得浅黄色纳米棒状氧化铈,记为CeO2-R[21]。
CeO2-F:该催化剂由沉淀法合成。室温剧烈搅拌下,称取2.78 g 硝酸铈和1.5 g 碳酸氢铵分别溶于100 mL 的水溶液中,使其充分溶解;经30 ℃下静置24 h 后,抽滤,洗涤,收集样品得到白色固体。再经80 ℃干燥12 h,450 ℃焙烧2 h,得到纳米片状氧化铈,记为CeO2-F[22]。
1.2 催化剂的表征
XRD 物相分析在日本理学D/max 2500 型衍射仪上进行X 射线衍射上进行,测试条件:CuKα射线源,管电压40 kV,管电流40 mA;扫描速率8(°)/min,5°-90°扫描。
氮物理吸附分析在贝士德3H-2000PS2 型物理吸附仪上进行。根据Barrett-Joyner-Halenda (BJH)方法分析材料的吸附-脱附等温线得到材料的比表面积、孔容、孔径分布等织构特性。
样品的微观形貌在日本电子株式会社JEM-2010F 型透射电子显微镜(TEM)上测定。测试条件:加速电压-200 kV。首先将样品置于无水乙醇中超声分散,滴于铜网,空气中风干后待测。
采用热电X 射线光电子能谱仪ESCALAB 250Xi 进行XPS 测试,以外来污染碳(284.8 eV)作为基准对测试结果进行荷电校准。使用XPS PEAK软件对获得的能谱图进行拟合解迭,对催化剂表面各类元素化学价态进行分析。
氢气程序升温还原(H2-TPR)在天津先权TP-5080 化学吸附仪上进行测试。首先,称取100 mg样品于石英管中,在氮气气氛(27 mL/min)下,升温至300 ℃预处理60 min。待样品管冷却至室温,改用5 % H2/N2混合气(27 mL/min)进行吹扫,并在此气氛下以10 ℃/min 的速率升温至900 ℃考察材料的还原性质。
氧气程序升温脱附测试(O2-TPD)在天津先权TP-5080 化学吸附仪上进行。氦气气氛(27 mL/min),样品装量50 mg,首先在120 ℃预处理60 min 以除去样品表面水分及其他挥发性组分。待样品管冷却至室温,改用5% O2/He 混合气(27 mL/min)进行吸附90 min,然后用氦气吹扫60 min 去除表面弱吸附的O2。随后以10 ℃/min 的速率升温至900 ℃考察材料对氧气的吸附-脱附性能。
拉曼光谱(Raman)谱图在英国雷尼绍inVia Reflex 激光拉曼光谱仪上采集得到,光源是波长为154.5 nm 的氩离子激光器,功率为20 mW,物镜为50 ×,光斑尺寸为10 μm。记录光谱0 - 4000 cm-1,整个测试在室温和常压下进行。
1.3 甲苯催化燃烧反应
以甲苯为探针分子进行的催化燃烧反应在实验室自主搭建的微型固定床-色谱装置进行,将混合气(20% O2/N2)通入冰水浴浸泡的甲苯中,通过鼓泡法将甲苯蒸汽携带进入反应器中,其中,甲苯蒸气质量浓度为1 g/L,尾气由气相色谱仪(GC-950,上海海欣色谱有限公司)分析,毛细管柱(SE-30,30 m × 0.32 mm × 0.25 μm)分离出产物,并采用氢火焰离子检测器(FID)记录信号。催化燃烧甲苯的反应是在常压下进行的,反应气体总流量为130 mL/min,气体空速为78000 mL/(g·L),反应温度为100-300 ℃。将100 mg 催化剂(40-80 目)与1 g石英砂混合均匀并置于微反应器的中部,利用石英砂良好的导热性能促进反应的传热、避免反应床层出现热点。t50和t90分别用来表示甲苯转化率为50%和90%对应的温度。
甲苯的转化率(x)的计算公式:

式中,[Toluene]in为反应进气中甲苯质量浓度;[Toluene]out为反应出气中甲苯质量浓度。
2 结果与讨论
2.1 催化剂形貌与织构性质
图1 为CeO2-A、CeO2-R 和CeO2-F 的不同放大倍率的TEM 照片。由图1 低倍率照片可以看出,CeO2-A 为气凝胶结构,由椭球形的纳米颗粒相互交联,形成立体网状结构,颗粒尺寸约为10 nm(图1((a)、(b)));CeO2-R 为完美的纳米棒结构,直径约为0.55 μm(图1((d)、(e));CeO2-F 由大小不同的纳米片堆叠而成,表面结构相对粗糙(图1((g)、(i))。从高分辨率透射电镜照片(图1((c)、(f)、(i))中可以清晰地看到晶格条纹,在CeO2-R 和CeO2-F 只有一种间距为0.312 nm 的晶格条纹;而CeO2-A 中同时出现0.270 和0.312 nm 的两种晶间距。因此,不同制备方法得到的氧化铈催化剂的形貌和暴露晶面存在明显差异,CeO2-R 和CeO2-F 催化剂主要暴露(111)晶面,而CeO2-A 催化剂主要暴露(111)和(100)两种晶面。
图2 为不同形貌CeO2样品的XRD 谱图。CeO2-A、CeO2-R 及CeO2-F 三种催化剂均在28.5°、33.1°、47.5°、56.3°及76.7°处出现明显的衍射峰,与萤石结构的CeO2(JCPDS No.43-1002)特征衍射峰一致。然而,由图2 可知,与CeO2-R、CeO2-F 相比,CeO2-A 中CeO2相的衍射峰发生明显宽化,说明晶粒尺寸较小,可能是由于晶格结构发生严重扭曲所引起的,可以暴露更多的氧空位[23]。另外,由Scherrer 公式计算得到的晶粒尺寸(见表1)可以看出,CeO2-A 比CeO2-R 和CeO2-F 的晶粒尺寸 明显降低,仅为10.6 nm[24]。

图1 不同制备方法得到的氧化铈TEM 照片Figure 1 TEM images of CeO2 prepared by different methods
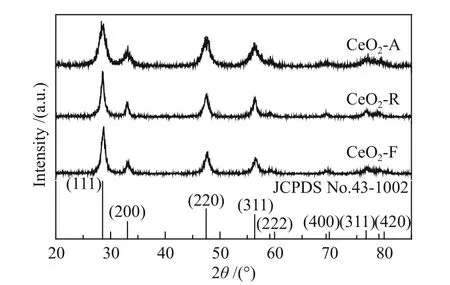
图2 不同形貌氧化铈催化剂的XRD 谱图Figure 2 XRD patterns of CeO2 catalysts with different morphologies
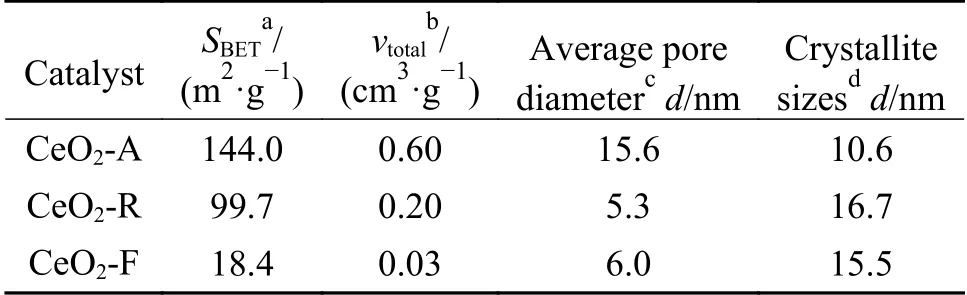
表1 不同形貌氧化铈催化剂的织构性质Table 1 Structural properties of CeO2 catalysts with different morphologies
图3 为CeO2-A、CeO2-R 及CeO2-F 催 化 剂 的N2吸附-脱附等温线及BJH 孔径分布曲线。由图3可知,不同形貌的CeO2催化剂均呈IV 型等温线,表明三种材料内部含有丰富的介孔结构[25,26]。从孔径分布曲线可以看出,CeO2-A 的孔道尺寸分布于3 nm 左右和大于5 nm 的两个区域,前者是由于干燥、焙烧过程中残留的有机分子挥发及前驱体盐热分解形成的孔道,后者可能属于CeO2纳米晶粒间形成的堆积孔[25]。CeO2-R 和CeO2-F 呈单孔分布状态,平均孔径约为5 nm。
表1 为CeO2-A、CeO2-R 及CeO2-F 催化剂的织构参数和晶粒尺寸。由表1 可知,比表面积与孔容的大小顺序一致、与晶粒尺寸顺序相反,这是由于增大孔容和降低粒径均有助于比表面积的提高。CeO2-A 比表面积(144.0 m2/g)明显高于CeO2-R(99.7 m2/g)及CeO2-F(18.4 m2/g),这可能由于CeO2-A 的制备工艺条件和物理化学过程有关,其高比表面积有利于反应物分子的吸附以及暴露更多的活性位点;同时,在三种催化剂中CeO2-A 具有最高的平均孔径和孔容,其丰富的孔道结构有利于反应过程中的传质,因此,认为采用溶胶-凝胶-超临界干燥法得到的多孔结构CeO2-A 更有助于甲苯的催化燃烧反应[27-29]。
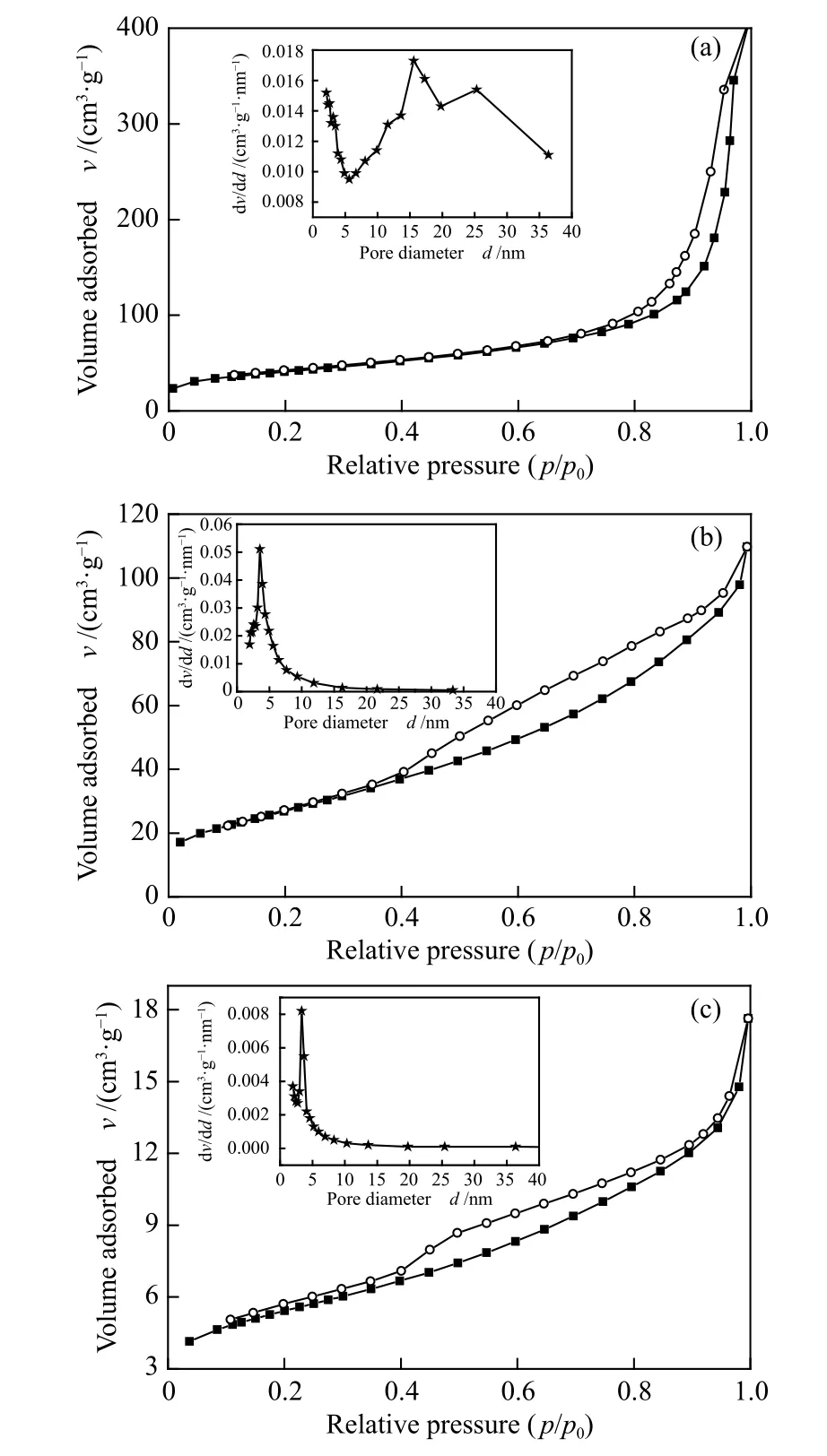
图3 不同形貌氧化铈催化剂的N2 吸附-脱附等温线和孔径分布Figure 3 N2 adsorption-desorption isotherms and pore size distributions of CeO2 catalysts with different morphologies
2.2 催化剂氧化还原性能
Wang 等[30,31]根据还原温度将氧化铈的还原峰归属于不同的氧物种,即100-400 ℃属于表面氧的还原,400-600 ℃为次表面氧的还原,高于600 ℃属于体相氧或晶格氧的还原,其中,催化剂表面更易被还原的氧物种是催化燃烧反应的活性氧物种。
不同形貌氧化铈催化剂的H2-TPR 谱图和相应数据分别见图4 和表2。图4 中,CeO2-A、CeO2-R 和CeO2-F 催化剂的还原峰可分为低温区(< 600 ℃)及高温区(> 600 ℃),分别对应于表面氧和晶格氧的还原;由图4 还可知,CeO2表面氧的起始还原温度随着催化剂形貌不同而不同:CeO2-A(281 ℃)<CeO2-R(313 ℃)< CeO2-F(328 ℃),表明CeO2形貌影响表面氧的活性,CeO2-A 表面氧起始还原温度最低,表明该催化剂具有更优异的低温还原活性[32]。另外,从表2 可以看出,相比CeO2-R 和CeO2-F,CeO2-A的低温还原峰面积最大,表明CeO2-A 具有更强的氧迁移能力以及更丰富的表面活性氧物种,还原性强,有助于提高催化燃烧性能[33]。
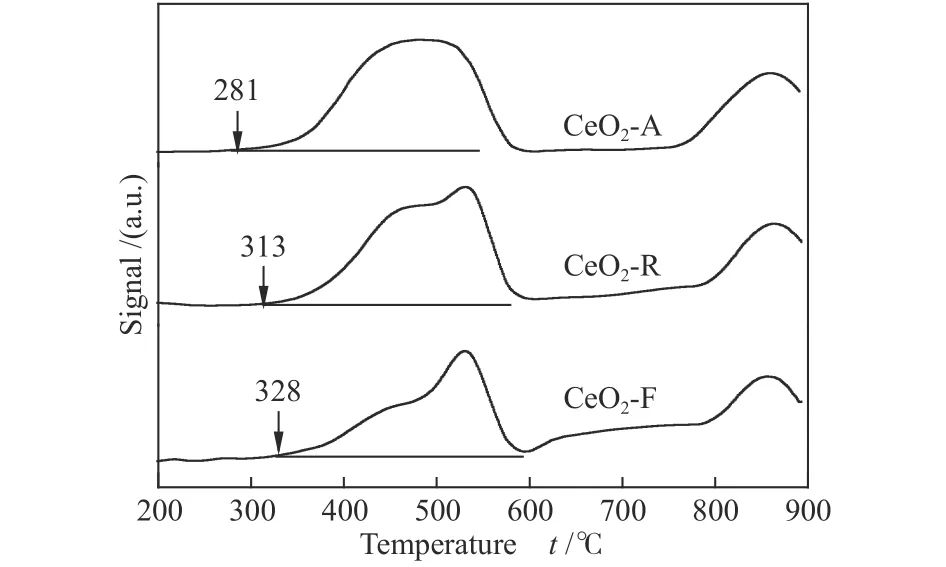
图4 不同形貌的氧化铈催化剂的H2-TPR 谱图Figure 4 H2-TPR spectra of CeO2 catalysts with different morphologies

表2 不同形貌的氧化铈催化剂TPR 表征Table 2 TPR results of CeO2 catalysts with different morphologies
采用O2-TPD 测试不同形貌氧化铈催化剂的氧气吸附-脱附行为、含氧物种类型及其移动性,见图5。Li 等[34]认为,一般在金属氧化物上氧物种主要分为四类:物理吸附分子氧O2(ad)、化学吸附氧(ad)、O-(ad)和晶格氧O2-(ad/lattice)。通常O2(ad)、(ad)和O-(ad)在100-500 ℃发生脱附,而O2-(ad/lattice)的脱附温度高于500 ℃[35]。其中,化学吸附的表面氧物种(ad)、O-(ad)被认为是催化燃烧反应的活性氧物种[36]。从图5 可以看出,CeO2-R、CeO2-F 仅在150 ℃出现脱附峰,其归属为O2(ad)的脱附;而CeO2-A 在150、350、463和663 ℃处出现四个较为明显的脱附峰,分别归属于O2(ad)、O2-(ad)、O-(ad)和O2-(ad/lattice)的脱附。换言之,与CeO2-R 和CeO2-F相比,CeO2-A 具有更丰富的活性氧物种O2-(ad)、O-(ad),有利于吸附的甲苯分子反应,加速甲苯催化燃烧反应;特别是高于500 ℃出现明显的O2-(ad/lattice)脱附峰,表明催化剂中晶格氧活动性强,氧物种具有很好的迁移性[37],与H2-TPR 结果一致,均说明三种形貌的催化剂中CeO2-A 具有最强的还原性能。
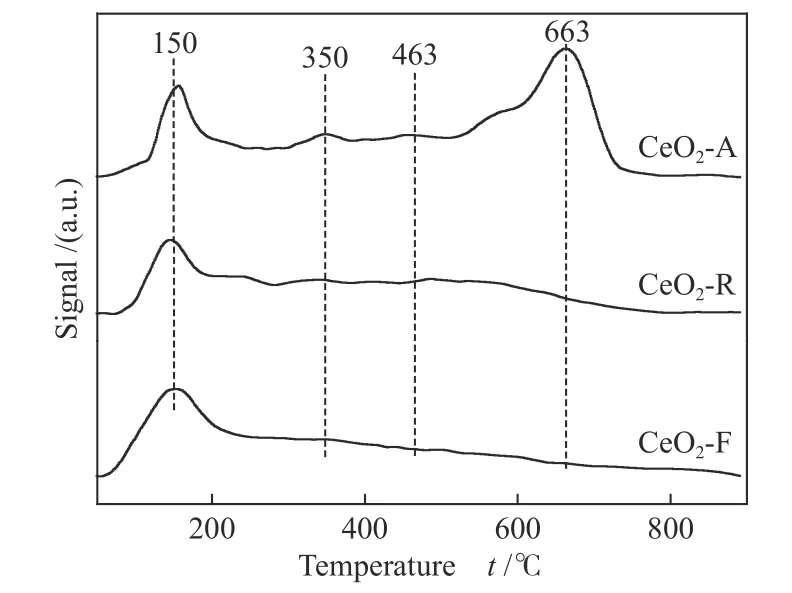
图5 不同形貌的氧化铈催化剂的O2-TPD 谱图Figure 5 O2-TPD spectra of CeO2 catalysts with different morphologies
2.3 催化剂表面化学性质
图6 为不同形貌氧化铈催化剂的拉曼光谱谱图。460 cm-1处有一个很强的拉曼振动峰,可归属为萤石结构的F2g特征振动,大约600 cm-1处的弱峰归属为氧空位的D 特征峰。通常,采用两种峰的强度比值ID/IF2g初步估计体相氧空位的含量,ID/IF2g值越大说明材料中氧空位越多[7,24,38]。图6还给出了不同形貌氧化铈的ID/IF2g值,可以看出CeO2-A 催化剂ID/IF2g值较大,说明不同的制备方法会对氧化铈样品的形貌产生极大的影响,进而促进氧空位的形成。
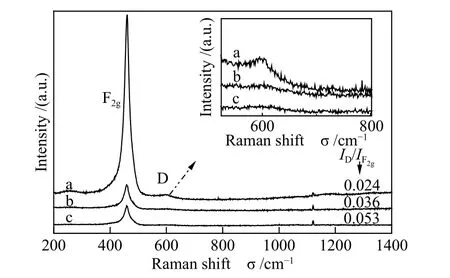
图6 不同形貌的氧化铈催化剂的拉曼光谱谱图Figure 6 Raman spectra of CeO2 with different morphologies a: CeO2-R; b: CeO2-F; c: CeO2-A
图7 为不同形貌氧化铈催化剂的XPS 谱图。根据Sakthivel 等[39]研究结果,对Ce 3dXPS 谱图(图7(a))进行去卷积分峰拟合可分为四对自旋轨道峰,分别标记为V、V1、V2和V3,如图6(a)所示。其中,V(882.1,900.6 eV)、V2(888.3,907.0 eV)及V3(898.1,916.3 eV)三对峰归属于Ce4+物种,而V1(884.5,903.0 eV)为Ce3+的特征峰。表3 为不同化学价态Ce 物种的定量分析结果,催化剂表面Ce3+/Ce 大小依次为CeO2-A > CeO2-R > CeO2-F。其中,CeO2-A 表面Ce3+/Ce 相对含量最高,意味着表面含有更加丰富的氧空位,有利于氧气的吸附与解离,从而促进催化燃烧性能[40]。
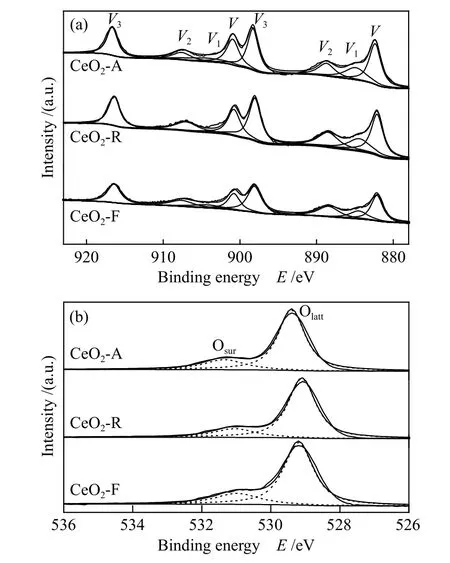
图7 不同形貌的氧化铈催化剂XPS 谱图Figure 7 XPS spectra of CeO2 catalysts with different morphologies

表3 不同形貌氧化铈催化剂的表面元素分析Table 3 Surface element analysis of CeO2 catalysts with different morphologies
图7(b)为不同形貌氧化铈的O 1sXPS 谱图。位于531.1-531.4 eV 处的特征峰归属于材料表面的晶格氧物种(记为Olatt),而529.3-529.7 eV 处的特征峰为表面吸附的氧物种(记为Osur)[41]。定量分析结果表明(表3),氧化铈样品表面氧物种相对含量(Osur/Olatt)有很大差异,由大到小依次为CeO2-A > CeO2-R > CeO2-F。由于在低温催化燃烧反应中,催化剂表面化学吸附氧物种是主要的活性氧,CeO2-A 表面丰富的化学吸附氧物种有利于提高其在低温区催化活性[42]。此外,催化剂表面Osur/Olatt比例可用于估算其表面氧缺陷的浓度,Osur/Olatt比值越大,氧缺陷浓度越高。由表3 可知,CeO2-A 表面氧空位浓度最高,氧吸附能力最强,这与拉曼和H2-TPR 结果一致。
2.4 活性评价
图8 为不同形貌氧化铈的催化燃烧甲苯活性评价结果,具体数值见表4。由图8 可知,三种催化剂转化率随反应温度升高而逐渐上升,且均在150 ℃甲苯开始发生缓慢转化。但与CeO2-F 相比,CeO2-A 和CeO2-R 的低温催化活性更好,在200 ℃时甲苯的转化率接近20%。随着反应温度继续升高,甲苯转化率迅速上升,CeO2-A 逐渐表现出更高的催化活性,t50和t90分别为223 及239 ℃。相比文献报道,沉淀法制备出的氧化铈纳米颗粒的t90为325 ℃[43],水热法制备出的氧化铈纳米棒的t90为300 ℃[44],CeO2-A 突显出较优异的催化性能,由此认为由于不同制备方法导致氧化铈样品的形貌、织构性质及表面化学性质存在明显差异,进而影响其催化燃烧性能。
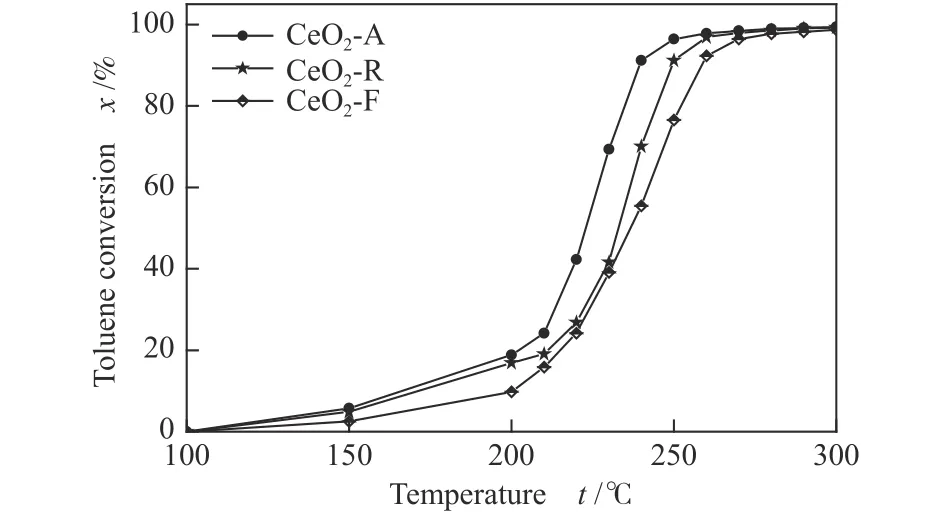
图8 不同形貌氧化铈催化剂对甲苯燃烧的催化性能Figure 8 Catalytic performance of CeO2 catalysts with different morphologies for toluene combustion reaction conditions: catalyst weight=100 mg, 1 g/L toluene,20 % O2/N2, total flow rate=130 mL/min and GHSV=78000 mL/(g·h)
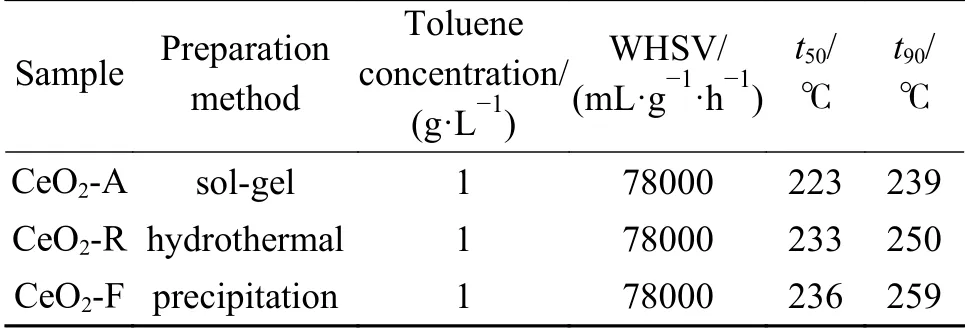
表4 不同形貌氧化铈催化剂对甲苯燃烧的活性评价Table 4 Catalytic activity of different morphologies CeO2 catalysts for oxidation of toluene
图9 为CeO2-A 的催化燃烧甲苯反应稳定性测试结果。在260 ℃反应长达50 h 的评价过程中,甲苯的转化率始终保持在98%以上,催化活性未出现明显降低,说明催化剂具有良好的稳定性,这可能归功于其结构的稳定性,提高催化剂抗失活能力。
通常过渡金属表面催化燃烧反应遵循Mars-Van Krevelen 反应机理[44],即催化剂表面活性氧物种与吸附的VOCs 分子之间反应,增强催化剂对反应物分子的吸附能力及自身的氧化还原循环能力,是提高催化剂催化活性的关键。
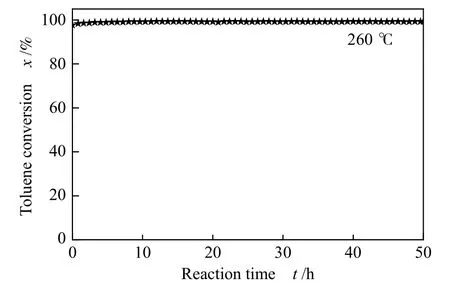
图9 CeO2-A 催化剂催化燃烧甲苯稳定性测试Figure 9 Long period test of catalytic oxidation of toluene over CeO2-A at 260 ℃
以上结果显示,不同制备方法得到的CeO2在物化性质上存在很大差异,进而影响其催化甲苯燃烧反应性能,CeO2-A 表现出最高的反应活性。首先,三种催化剂的活性、比表面积和氧空位浓度顺序一致,均为CeO2-A > CeO2-R > CeO2-F,表明CeO2-A 较大的比表面积,促进其表面更多氧空位的暴露(Osur/Olatt= 0.25),有利于反应物分子的吸附,加速甲苯燃烧反应进行;并且,CeO2-A 同时暴露(100)和(111)活性晶面,而CeO2-R、CeO2-F 仅暴露(111)一种晶面,不同晶面形成氧空位的能垒顺序为:(110)< (100) < (111)[9,45],因而具有高暴露活性晶面的CeO2-A 比其他两种形貌的催化剂容易形成更多的氧空位。还有值得注意的是,CeO2的(100)晶面为极性晶面,最外层为密堆积O 原子;而CeO2的(111)晶面是非极性晶面,由化学计量的Ce4+和O 原子组成,CeO2-A 的(100)晶面可能更有利于极性分子甲苯的吸附[46]。所以,除具有大的比表面积外,高暴露活性晶面也是使CeO2-A 表面氧空位浓度增加,有利于甲苯吸附,进一步提高活性的重要因素。此外,三种形貌氧化铈的还原能力存在显著差异,H2-TPR 结果表明,CeO2-A 的还原性最强,CeO2-R 其次、CeO2-F 最弱;O2-TPD 结果进一步证实晶格氧活动性顺序与其一致,因此,CeO2-A 的自身的氧化还原循环能力最强,有利于产生更高浓度的活性氧物种,促进反应物分子的吸附解离,进而提高催化反应活性。
综上所述,大比表面积和高暴露活性晶面均可促进氧化铈表面氧空位浓度的提高,有利于反应物分子的吸附;晶格氧活动性的增强,有利于氧化铈氧化还原循环能力的提高;氧空位浓度和氧化还原循环能力的提高是CeO2-A 催化剂在催化燃烧甲苯反应中表现出优异性能的重要原因。
3 结 论
分别采用溶胶-凝胶-超临界干燥法、水热合成法及沉淀法分别制备了不同形貌的CeO2-A、CeO2-R和CeO2-F 催化剂,在甲苯的催化燃烧反应中均表现出良好的催化活性,甲苯完全转化温度(t90)分别为239、250 和259 ℃,CeO2-A 的催化活性最佳。
CeO2-A 具有最高的比表面积,有利于更多吸附位点的暴露和反应物分子的吸附;CeO2-R 和CeO2-F 仅暴露(111)晶面,而CeO2-A 同时暴露(100)和(111)晶面,晶面结构的不同造成氧空位浓度和对甲苯分子吸附能力的不同,使CeO2-A 比其他两种催化剂具有的表面氧空位浓度更高和对甲苯分子的吸附能力更强;CeO2-A 催化剂中晶格氧活动性强,促进自身氧化还原循环能力提高。因此,采用不同方法制备的三种形貌CeO2催化剂中,CeO2-A 比CeO2-R 和CeO2-F 表现出更优异的催化燃烧甲苯反应性能,溶胶-凝胶-超临界干燥法更有利于制得催化高性能催化剂样品,主要归因于比表面积大、暴露(111)和(100)活性晶面及晶格氧迁移性强。
猜你喜欢
能源工程(2021年2期)2021-07-21
校园英语·月末(2019年11期)2019-09-10
分析化学(2019年3期)2019-03-30
作文中学版(2018年1期)2018-11-28
当代陕西(2018年9期)2018-08-29
科学与财富(2017年27期)2017-10-17
科技资讯(2017年24期)2017-09-15
哈尔滨理工大学学报(2015年5期)2016-01-19
读者欣赏(2014年6期)2014-07-03
