
硝基咪唑在TiO2晶面的吸附性质和降解机理研究*
2021-02-23 07:40谭若兰王译伟郭建敏
广州化工 2021年3期
何 扬,谭若兰,张 春,王译伟,郭建敏
(1 西南医科大学药学院,四川 泸州 646000;2 西南医科大学基础医学院,四川 泸州 646000)
硝基咪唑类抗生素是一类具有广谱活性的硝基杂环化合物,临床应用普遍。它们在一些废水处理厂甚至淡水环境中被检测出来[1],虽然浓度很低,但对环境潜在的威胁依然存在。吸附法和生物法[2-4]是当前已经普及的降解方法。然而吸附法仅是简单地将污染物从水相置换到固相,却不能真正将其降解[5]。生物法需要较长时间,多种因素将会造成干扰,降解效率因环境而异[6-7]。物理法中的絮凝和离心操作往往也会造成二次污染。氧化尽管也是一种不错的选择,但其降解过程中的产物可能产生更大的毒性[8]。 近年来,光辅助TiO2催化法作为一种高级的氧化手段,可用于处理抗生素污染的废水,因其反应条件温和、简单易行等优点在降解污染物方面颇受关注[9]。一些研究表明,TiO2光催化可用于降解多种硝基咪唑类抗生素[10]。因此对硝基咪唑类抗生素的降解机理和吸附特性以及光辅助对其催化降解的影响进行概述具有重要意义。
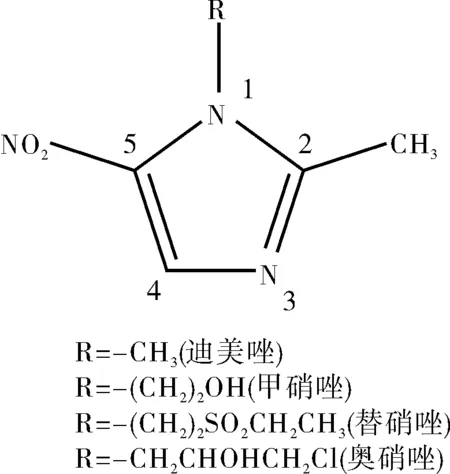
图1 常用硝基咪唑类结构式
1 TiO2表面构型简介和程序操作
1.1 TiO2表面构型简介
TiO2表面通过量子化学法被人工构建了两个模型系统,即锐钛矿型TiO2(101)表面和TiO2(001)表面,基于板厚对表面能影响的初步研究[20],发现TiO2(101)和TiO2(001)两种表面可以充分模拟TiO2表面[21-22]。如图2,图3所示,TiO2(101)表面有两种Ti(钛)原子和两种O(氧)原子,它们分别是Ti(5)和Ti(6),O(2)和O(3),且表面为三层模型,通常由1×3×3超晶胞模拟。TiO2(001)表面和TiO2(101)表面的原子类型一样,但TiO2(001)的Ti(6)不存在表面层,而是偏向内层,且表面为单层,通常由3×3×1超单胞模拟。且经证明Ti(5)和O(2)原子比Ti(6)和O(3)原子更为活跃[13-16]。为了避免平板与其周期图像的相互作用,在垂直方向上增加了15°的真空空间。在中性水溶液中,在宇宙力场作用下,按1 g/cm3的密度分别在TiO2(101)和(001)表面上分别加入48和69个H2O分子[13-19]。在酸性(碱性)条件下,H2O分子被HCl(NaOH)取代[16]。

图2 TiO2(101)(a~b)和(001)(c~d)晶面的侧视图和俯视图[13]

图3 TiO2(101)(a)和(001)(b)晶体立体表面结构[14]
1.2 操作程序
将硝唑咪唑类分子分别置于TiO2(101)和(001)表面,为了消除表面与该类分子间较强地相互作用,将它们之间设置一定距离。在LAMMPS程序下引入ReaxFF(反作用力场)和NVE[13-19](粒子、体积、总能量都保持恒定的微正则系统)或NVT(粒子、体积、温度都保持不变的微正则系统)来进行分子动力学计算[23-24]。在LAMMPS松弛结果的基础上,利用DFT(密度泛函理论)选择局部最小结构进行进一步优化。之后再转移到Materials Studio程序中,用DMol3板块进行DFT计算进行结构优化,通过比较各自吸附能力大小、吸附距离以及键长吸附前后改变情况来确定最佳吸附位点以及降解位点,在最终确定其最佳吸附模型和降解机理。并采用COSMO[25-26]模型,分别以水、酸、碱为溶剂,研究反应的溶剂化效应[13-19]。
2 真空、中性水溶剂下的硝基咪唑分子在TiO2表面的吸附特性
2.1 真空中的吸附特性
经DFT测定,真空条件下迪美唑、甲硝唑、奥硝唑、替硝唑在TiO2(101)和(001)表面均有5种不同稳定吸附构型,且四种同类抗生素与TiO2(101)和(001)的表面吸附均存在多个位点,同时,咪唑环C(5)的硝基、N(1)上羟基或磺酰基的O原子都能吸附在TiO2表面的Ti(5)原子上,并且环上C(2)甲基和N(1)支链上的H原子都可TiO2的O(2)原子形成氢键[13-17]。Zhang等[27]研究了染料与TiO2的界面吸附,发现氢键可使染料在TiO2表面聚集的稳定性增加。Li等[28]表明,纳米复合支架的稳定性也被PVP与TiO2之间的氢键增强。Chang等[29]研究表明,HNO3和TiO2之间的氢键可提升吸附能量和吸附构型的稳定性。结果均表明氢键的形成可以提高吸附构型的稳定性。
TiO2(101)与和四种硝基咪唑分子结合的最稳定的吸附构型:N(3)原子吸附在Ti(5)上,且C(2)上甲基的部分H原子与TiO2(101)表面的O(2)原子形成氢键。此构型下,由于N(3)原子与TiO2(101)表面的Ti(5)原子相互作用,三者的N(1)-C(2)的共价键键长通常都会增大,且N(1)-C(2)键的减弱会让羟基自由基的攻击更有利[13-17]。
TiO2(001)与四种硝基咪唑类分子结合的最稳定的吸附构型:咪唑环上的N(3)吸附在TiO2(001)表面上的Ti(5)上,且四者甲基上的部分氢原子与TiO2(001)表面的O(2)原子形成氢键[13-17]。与吸附催化剂前的结构相比,迪美唑、甲硝唑和替硝唑均是引起N(1)-C(2)和N(3)-C(4)键相应的增长,且N(1)-C(2)键的减弱同样也让羟基自由基的进攻更有利[13-14,17]。而奥硝唑则是因为C(2)-N(3)键的增长使其更容易被羟基自由基进攻[15]。
2.2 中性溶剂下的吸附特性
四种硝基咪唑类分子在中性溶剂的TiO2表面上仍为多点吸附,且水溶剂模型的加入后,水分子中的氢原子与氧原子在晶体表面形成氢键,使吸附更加牢固[13-15]。
奥硝唑、迪美唑与TiO2(101)在中性溶液下最稳定的吸附构型与真空条件下相似[15,17]。而甲硝唑最稳定的吸附构型:甲硝唑上的羟基O原子吸附在TiO2(101)表面的Ti(5)上。同时,其环上的羟基H原子与TiO2(101)表面的O(3)形成氢键且其N(1)支链上和甲基上的H原子与TiO2(101)表面的O(2)形成氢键[13-17]。替硝唑最稳定的吸附构型:其磺酰氧原子吸附在TiO2(101)表面的Ti(5)上,N(1)支链上的H原子和TiO2(101)表面的O(2)形成氢键[14]。
对于(001)表面,迪美唑、奥硝唑、甲硝唑最稳定的吸附构型与在真空的吸附特性相似[13,15,17]。而TiO2(001)表面虽存在替硝唑最稳定的构型,但具体的共吸附方式缺乏报道,只知此构型在水溶下的N(1)-C(2)和N(3)-C(4)键比吸附前有所增长[14]。
综上所述,在真空和中性环境下,可推测出大部分硝基咪唑分子最稳定的吸附构型通常为:其N(3)原子与TiO2的Ti(5)的互相吸附。该构型吸附能往往最大,吸附均使碳氮键长变长,有利于自由基的进攻和开环反应。同时也可推测出大部分硝基咪唑类分子与水分子的吸附过程中会产生较强的作用力,这也使得吸附能较真空条件下有所增加,吸附构型也相对更为稳定。如Schneider等[30]研究了卟啉在TiO2酸酶颗粒上的吸附和Mendive等[31]研究了水溶液中草酸根在锐钛矿(100)和金红石(110)上的吸附,都得到了相似的结论。在酸、碱性条件下,由于硝基咪唑分子与TiO2(101)和(001)面的共吸附相关报道很少,所以暂不明确其具体的吸附特性。
3 硝基咪唑分子在TiO2表面催化降解反应机理
如图3、图4所示,反应路线I为R(反应物) → TS1(过渡态1)→M1(中间产物1)→TS2(过渡态2)→P(最终产物)。首先吸附在催化剂上的水被空穴氧化成·HOH,·H去进攻C(2),导致C(2)-N(1)被破坏,于是在C(2)处形成烯醇结构,最后再通过四元环结构的过渡态,羟基上的H转移到咪唑环的N(3)上,形成最终产物。
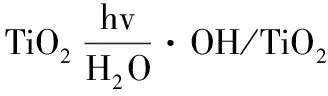
图3 光照下水在TiO2表面上的反应

图4 硝基咪唑的开环降解的两条反应路径
反应路线II为R→TS3→ M2→TS4→P。首先羟基上的H原子通过四元环过渡态转移到咪唑环的N(3)上,然后造成C(2)-N(1)键断裂并形成最终产物。
奥硝唑(NVT系统下)、迪美唑、甲硝唑和替硝唑(NVE系统下)在TiO2(101)和(001)表面上的水溶剂模型条件下开环反应机理与真空相似,且都是按照图4的两条路径进行,只是水溶液条件下的吸附过程中的氢键效应会使共吸附更加稳定[13-15,17]。甲硝唑分子在TiO2(101)表面的开环反应活化能比TiO2(001)表面低,且通道II控制步骤的活化能比反应通道I中开环步骤的活化能大,因此,甲硝唑分子在TiO2(101)表面以通道I的方式进行开环为最佳反应路径[13],该路径可使咪唑环更快地被进行催化开环反应。同样,奥硝唑在TiO2(001)表面以开环反应路径I进行最佳[15]。迪美唑和奥硝唑的反应路径一样,只是开环可能需要加热[17]。而替硝唑分子在TiO2(101)表面以通道II的方式进行开环反应更为容易[14],其他硝基咪唑具体的反应途径还有待研究。

4 降解的影响因素
4.1 光源的影响
光源是光催化降解作用必不可少的条件之一。在真空时和中性溶剂时,TiO2的能隙都会通过吸附而缩小[13,15],可使其激发的光谱范围变得更广。Tan等[16]研究证明,真空,中性溶剂,酸性,碱性条件下的五种结构的能隙值均在可见光范围(可见光范围约为1.7~3.1 eV)[33-34],证明可见光可有效的驱动奥硝唑在TiO2表面的降解。Wang等[13]、Qin等[14]也分别证明了可见光可辅助TiO2催化降解甲硝唑和替硝唑。Qin等[18]也证明TiO2(101)在可见光下有利于催化降解迪美唑。Farzakia等[11]使用UV/TiO2对甲硝唑进行降解对比实验,在pH、TiO2和甲硝唑初始浓度一定时,反应3 h,对甲硝唑的去除率可达到97.61%。陈冬梅等[12]实验证明了增加光强度,可有效加大迪美唑的降解率。
当TiO2光催化技术对其他类的抗生素和药物进行处理时,同样具有良好的效果。莫西沙星可有效地被TiO2所降解[35]。高乃云等[36]发现水中大部分的磺胺甲恶唑能被除去。王佳婕等[37]用沸石和TiO2的复合材料在紫外光下进行光催化氧化对乙酰氨基酚,3 h后可达到很高的去除率。
4.2 其他因素的影响
4.2.1 pH的影响
Farzakia等[11]通过实验证明了在pH=7时,甲硝唑降解效果最好。Tan等[16]证明TiO2表面的酸性环境有利于奥硝唑分子的吸附,而碱性不利于吸附。陈冬梅等[12]通过实验证明了pH值的升高会伴随更多·OH的产生,对迪美唑的降解效果最好。由此可知,溶液pH对TiO2光氧化能力的影响较为复杂,且不同操作或药物条件下所进行的光降解有着不同的最佳pH值。
4.2.2 TiO2剂量的影响
经实验证明[12,15,37],在一定范围类,光催化剂用量适当地增加可提高对硝基咪唑类抗生素的降解效率。随着用量继续加大,TiO2会发生团聚集,紫外光将会被散射,造成光透过率变差,从而促使反应速率下降。
4.2.3 初始药物浓度的影响
Farzakia等[11]实验证明了随着甲硝唑初始浓度的增加,光催化降解效率降低。推测可能是随着甲硝唑浓度的持续增加,MNZ和其降解中间体可能占据了TiO2表面的大部分,从而对·OH的利用和TiO2表面价带上的正空穴产生了负面影响。并且浓度进一步增大还可能会引起内过滤效应(药物浓度的增加造成其本身对紫外光的吸收加大),这种效应会使极少的光子能够到达TiO2表面[38]。陈冬梅等[11]也通过实验证明迪美唑的初始浓度对其降解效率为先增加后减少,其他研究人员也报告了类似的结果[39-41],因此可推测硝基咪唑类抗生素类的初始浓度对其本身降解效率的影响一般为先增后减。
5 结 语
本文通过应用DFT,着重总结了真空和中性条件下大部分硝基咪唑抗生素在TiO2晶体面的最佳吸附特性和降解机理,但酸、碱性条件下两者共吸附的相关报道较少,有待深入挖掘。归纳了甲硝唑、迪美唑等硝基咪唑药物在TiO2晶体面上被催化降解的相关影响因素,发现可见光可被充分利用,且光源是不可或缺的条件之一。与此同时,不同的pH环境、药物的初始浓度、TiO2剂量等也会对其造成重要影响。
DFT计算法与实验探究法相比,虽真实性存在一定偏差,但也为其他的硝基咪唑类抗生素更深入的研究提供了新的思路,两者方法互相结合,将会为有更广阔的应用前景。
猜你喜欢
农产品加工(2022年14期)2022-11-17
中学生学习报(2021年20期)2021-11-24
鞍钢技术(2021年3期)2021-06-11
化工管理(2021年7期)2021-05-13
四川畜牧兽医(2020年7期)2020-07-20
老年医学与保健(2017年6期)2017-02-06
中国农资(2016年1期)2016-12-01
中国卫生标准管理(2015年25期)2016-01-14
中国卫生标准管理(2015年24期)2016-01-14
化工进展(2015年3期)2015-11-11
