
世界卫生组织中枢神经系统肿瘤分类的演变:1979-2021年
2021-02-10 06:20杨学军江涛陈忠平于士柱
中国现代神经疾病杂志 2021年9期
杨学军 江涛 陈忠平 于士柱
神经系统肿瘤的病理学研究始于19世纪。最初的神经肿瘤病理学研究受限于研究手段,仅能够对肿瘤进行肉眼观察(Cruveihier,1829年),病理组织标本往往是尸检材料,这种直观的大体病理学揭开了神经病理学研究的序幕。高质量显微镜的应用促进了细胞的发现(Schleiden,1838年;Schwan,1839年),将病理学家的视觉从宏观层次拓展至微观层次,为从组织学角度辨别神经肿瘤奠定了基础。19世纪中下叶,Virchow对颅内肿瘤的病理学研究做出重要贡献,1846年他首先提出“胶质瘤”的概念,并把发生于硬脑膜、具有“砂粒体”结构的肿瘤命名为“砂粒瘤”,明确指出应该与硬脑膜“肉瘤”区分开来[1-3]。
20世纪初,神经外科在欧洲发展起来,手术入路、技术和安全性的提高,为病理学家提供不同部位、不同组织学特点的颅内肿瘤标本,神经病理学研究开始与患者临床特点、治疗反应和预后结合起来。1926年,Bailey和Cushing首次提出神经上皮组织肿瘤的系统分类,并依据胚胎残留学说初步分为14种类型,即髓上皮瘤、髓母细胞瘤、松果体母细胞瘤、松果体细胞瘤、室管膜母细胞瘤、室管膜瘤、神经上皮瘤、极性胶质(海绵)母细胞瘤、星形母细胞瘤、星形细胞瘤、少突胶质细胞瘤、神经母细胞瘤、神经节细胞瘤、脉络丛乳头状瘤,并提出“肿瘤分级”的概念,将颅内肿瘤病理学特点与患者预后相关联[1-3]。他们认为,神经系统胚胎发育过程中如果出现细胞分化障碍,胚胎残留细胞出生后遂形成肿瘤;成熟细胞的肿瘤属相对良性肿瘤,由胚胎或幼稚细胞组成的肿瘤属恶性肿瘤[1-3]。尽管由于科学发展的局限,这一理论在细胞分化和肿瘤发生方面存在错误和缺陷,但仍对神经肿瘤学的发展做出了重要贡献,是当代神经上皮组织肿瘤分类的基础,许多肿瘤实体命名仍沿用至今[1-3]。
20世纪中后期,许多学者在脑肿瘤分类方面做出重要贡献,如Penfield、Hortega、Kernohan和Sayre、Zülch、Rusell和Rubinstein,但是这些学者在分类概念、组织学标准和分类方法方面并不完全相同,不同的分类系统用于不同的国家和地区,如Kernohan系统主要在英语国家应用,Hortega系统主要在葡萄牙语和西班牙语国家应用。我国病理学家和神经科学工作者也提出自己的分类方法如黄克维、张福林、黄文清、吴在东等以及北京神经外科研究所的中枢神经系统肿瘤分类。从世界范围看,对中枢神经系统肿瘤的分类,多数学者秉承Bailey-Cushing、Kernohan和Zülch等的学术思想。可想而知,不同的中枢神经系统肿瘤分类使神经肿瘤学研究与诊治结论之间的相互比较进一步复杂化,也给文献阅读和学术交流带来困难[1-3]。
基于此背景,世界卫生组织(WHO)自1956年开始致力于建立一套可以被全世界接受和应用的肿瘤分类和分级系统。自1979年在瑞士苏黎世大学Zülch教授领导下出版WHO中枢神经系统肿瘤分类(第一版,以下简称第一版分类)以来,迄今已出版5版共6个版本(表1)[4-9]。由于WHO中枢神经系统肿瘤分类系列书籍的封面均为蓝色基调,常简称为“蓝皮书”。各版WHO中枢神经系统肿瘤分类需确定和调整的内容包括中枢神经系统肿瘤分类的框架、肿瘤类型/亚型(命名法、归属、增加、暂定或删除)、WHO分级、肿瘤分类与临床诊治和预后的对应关系。自2000年WHO中枢神经系统肿瘤分类(第三版,以下简称第三版分类)开始,“蓝皮书”从“小册子”发展至几百页的“书”,对各种类型肿瘤病理学特点进行精确注释,丰富了分子生物学和分子遗传学等信息,还简要描述了流行病学、临床症状与体征、影像学、结局和预测因素[10]。
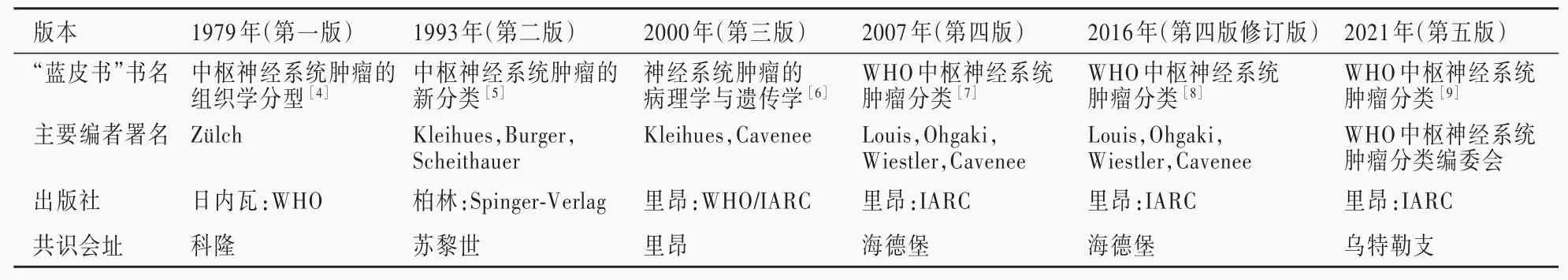
表1 WHO中枢神经系统肿瘤分类出版一览表Table 1. The synoptic table of published WHO Classification of Tumors of the Central Nervous Systerm
自1926年Bailey和Cushing首次提出神经上皮组织肿瘤的系统分类以来,“组织发生”概念主导中枢神经系统肿瘤分类近1个世纪,即通过与所推测判定的发生细胞组织学相似性和细胞分化水平进行分类,技术上主要依靠显微镜下HE染色、谱系相关蛋白免疫组化染色和超微结构观察。以组织学为基础的WHO肿瘤分类和分级系统作为“金标准”,在中枢神经系统肿瘤的诊断与治疗中发挥重要作用。然而,组织学诊断可能出现模棱两可或不同观察者之间存有差异;根据组织学标准诊断的同一肿瘤类型也存在生物学行为、临床特点、治疗反应和结局的不同[11]。尽管中枢神经系统肿瘤的分子生物学和分子遗传学研究成果自第三版开始写入“蓝皮书”,2007年WHO中枢神经系统肿瘤分类(第四版,以下简称第四版分类)陈述某些肿瘤发生发展的分子特征,但是由于条件尚不成熟,仅在传统组织学确定的分类框架内作为临床预后或预计参考指标。2016年,WHO首次在组织学诊断的基础上附加分子特征,尝试对部分肿瘤如弥漫性胶质瘤、髓母细胞瘤进行整合诊断,并定为WHO中枢神经系统肿瘤分类第四版修订版(以下简称第四版修订版)。由于分子肿瘤学的发展远超WHO肿瘤分类更新的步伐,一些有前景的生物学标志物和新药物靶点的发现,助推WHO中枢神经系统肿瘤分类更新的进程[11]。中枢神经系统肿瘤分子信息与分类实践联盟-非WHO官方组织(cIMPACT-NOW)的建立是一种新的机制,可及时对临床相关研究中可能应用于临床实践的知识进行评估,弥补临床与科研之间存在的知识差距,澄清困惑,为新版分类提供建议和讨论[12]。经过cIMPACT-NOW的7次更新,2021年WHO中枢神经系统肿瘤分类(第五版,以下简称新版肿瘤分类)在胶质瘤、胶质神经元肿瘤和神经元肿瘤、胚胎性肿瘤中更广泛地采用联合组织学表型和分子特征的整合性命名。
一、中枢神经系统肿瘤分类框架的演变
第一版分类将中枢神经系统肿瘤分为12大类,即神经上皮组织肿瘤、神经鞘细胞肿瘤、脑膜及相关组织肿瘤、原发性恶性淋巴瘤、血管起源的肿瘤、生殖细胞肿瘤、其他畸形性肿瘤和类肿瘤病变、血管畸形、垂体前叶肿瘤、局部延伸性肿瘤、转移性肿瘤、未分类肿瘤[4]。1993年WHO中枢神经系统肿瘤分类(第二版,以下简称第二版分类)删除第一版中血管起源的肿瘤、血管畸形及个别肿瘤的重复命名,经调整和补充而缩减为10大类,即神经上皮组织肿瘤、颅神经和脊神经的肿瘤、脑膜肿瘤、淋巴瘤和造血组织肿瘤、生殖细胞肿瘤、囊肿和类肿瘤病变、鞍区肿瘤、局部延伸性肿瘤、转移性肿瘤、未分类肿瘤[5]。第三版分类开始尝试采用肿瘤性疾病国际分类(ICD-O)和医学系统命名法(SNOMED)编码和标识神经系统肿瘤,删除囊肿和类肿瘤病变、局部延伸性肿瘤,取消未分类肿瘤及肿瘤亚型中“其他”条目,将开放式肿瘤分类改为封闭式神经系统肿瘤分类,重新归类为7大类,即神经上皮组织肿瘤、周围神经肿瘤(首次包括周围神经系统肿瘤)、脑膜肿瘤、淋巴瘤和造血组织肿瘤、生殖细胞肿瘤、鞍区肿瘤、转移性肿瘤[6]。第四版分类仍采用组织学分类原则,继续分为神经上皮组织肿瘤、颅神经和椎旁神经肿瘤、脑膜肿瘤、淋巴瘤和造血组织肿瘤、生殖细胞肿瘤、鞍区肿瘤、转移性肿瘤共7大类,并取消“来源未定”条目[7]。第四版修订版首次构筑分子时代中枢神经系统肿瘤诊断的结构框架,共分为17大类,与第四版分类一级条目的对应关系为,第四版修订版条目1~8对应第四版分类的神经上皮组织肿瘤、条目10~12对应脑膜肿瘤、条目13~14对应淋巴瘤和造血组织肿瘤、其他一级条目无变化,其中,神经上皮组织肿瘤分类结构的调整主要体现在弥漫性胶质瘤、髓母细胞瘤、胚胎性肿瘤[8]。新版肿瘤分类将中枢神经系统肿瘤分为12类,即胶质瘤、胶质神经元肿瘤和神经元肿瘤(成人型弥漫性胶质瘤、儿童型弥漫性低级别胶质瘤、儿童型弥漫性高级别胶质瘤、局限性星形细胞胶质瘤、胶质神经元和神经元肿瘤、室管膜肿瘤)、脉络丛肿瘤、胚胎性肿瘤、松果体肿瘤、颅神经和椎旁神经肿瘤、脑膜瘤、间叶性非脑膜皮肿瘤、黑色素细胞肿瘤、血液和淋巴肿瘤、生殖细胞肿瘤、鞍区肿瘤、中枢神经系统转移性肿瘤,与第四版及其修订版的对应关系参见表2[9]。

表2 2007-2021年WHO中枢神经系统肿瘤分类的框架对比Table 2. The category frame comparison of WHO Classification of Tumors of the Central Nervous Systerm from 2007 to 2021
二、中枢神经系统肿瘤类型(实体)及亚型(变型)的演变
光学显微镜、电子显微镜、免疫组化染色的应用使病理学诊断手段得以丰富,显著提高诊断的精确性。每版WHO肿瘤分类,尤其是前几版均会废除上一版有争议、但经分子生物学证实不存在的肿瘤命名,并调整肿瘤实体的类别归属,以及增加新发现的中枢神经系统肿瘤。即使在以组织学形态为基础的情况下,各版肿瘤分类新增肿瘤实体也是有原则的:必须有来自不同医疗机构的2份及以上的肿瘤报告,不仅具有独特的病理学表现,还必须在发病部位、年龄和生物学行为方面具有特征[3]。新的肿瘤变型(variant)的增加应符合以下原则:首先属于已确定的肿瘤实体,但在组织学上又具有可靠的自身识别特征,同时与临床预后具有相关性。然而不均一的组织学表现和多样的组织学形态为肿瘤所常见,很多独特的组织学表现在临床行为和基因型上并不具有特殊之处,因此并非每个可识别的组织学表现均可以被指定为一种实体/变型(entity/variant)[3]。应以发展和动态眼光看待这一问题,即使暂时认为可能仅是组织学异向分化形式,也不排除随着临床和随访资料的积累以及独特分子特征的发现,有可能在将来被确认为新的肿瘤实体/变型。
第四版修订版在争取不打乱现有临床处理以及临床与流行病学对应关系的前提下,突破肿瘤分类和病理诊断完全依赖显微镜的局限,将分子诊断指标引入中枢神经系统肿瘤分类,主要在弥漫性胶质瘤、髓母细胞瘤、胚胎性肿瘤中进行尝试[8,11]。向新版肿瘤分类的过渡阶段,cIMPACT-NOW以7次更新的形式发布下述提议(表3)[13-20]:(1)与其他非中枢神经系统肿瘤的术语一致,采用肿瘤“类型/亚型(type/subtype)”对应替代既往各版肿瘤分类中的“实体/变型”[13]。(2)与其他非中枢神经系统肿瘤一致,在肿瘤类型内进行WHO分级,并用阿拉伯数字(WHO 1~4级)替代罗马字母(WHOⅠ~Ⅳ级)[13]。(3)未行辅助分子检测或结果无法解释,导致仍基于组织学形态诊断时,标识“NOS”;进行分子检测,但肿瘤类型、组织学形态、免疫表型和基因型与WHO标准肿瘤类型不匹配,而仅做描述性诊断时,标识“NEC”[14]。(4)IDH野生型和IDH突变型弥漫性胶质瘤是不同的疾病,具有不同的分子特征谱,临床预后亦不同,胶质母细胞瘤不再用于IDH突变型肿瘤的诊断;启用弥漫性星形细胞瘤,IDH野生型,具有胶质母细胞瘤分子特征,CNS WHO 4级和星形细胞瘤,IDH突变型,CNS WHO 4级的诊断[15-16]。(5)H3突变型弥漫性胶质瘤体现出表观遗传学在肿瘤分类中的作用,弥漫性中线胶质瘤,H3 K27M突变型的诊断须符合弥漫性生长、位于中线部位、胶质瘤组织学形态和H3 K27M突变的全部特征,并建议新实体——弥漫性胶质瘤,H3.3 G34突变型[17]。(6)儿童型胶质瘤/胶质神经元肿瘤的分类建议采用矩阵匹配模式,即在矩阵表格中分别列出组织学信息和基因型信息,通过组织学信息与基因型信息的整合定义肿瘤类型,同时根据其他分层信息进行诊断[18]。(7)室管膜瘤根据解剖部位和分子表型/相关基因改变进行分类,以反映其生物学特征。脊髓室管膜瘤,MYCN扩增型作为脊髓室管膜瘤的独立亚型,预后不良。黏液乳头状型室管膜瘤升至CNS WHO 2级[19]。
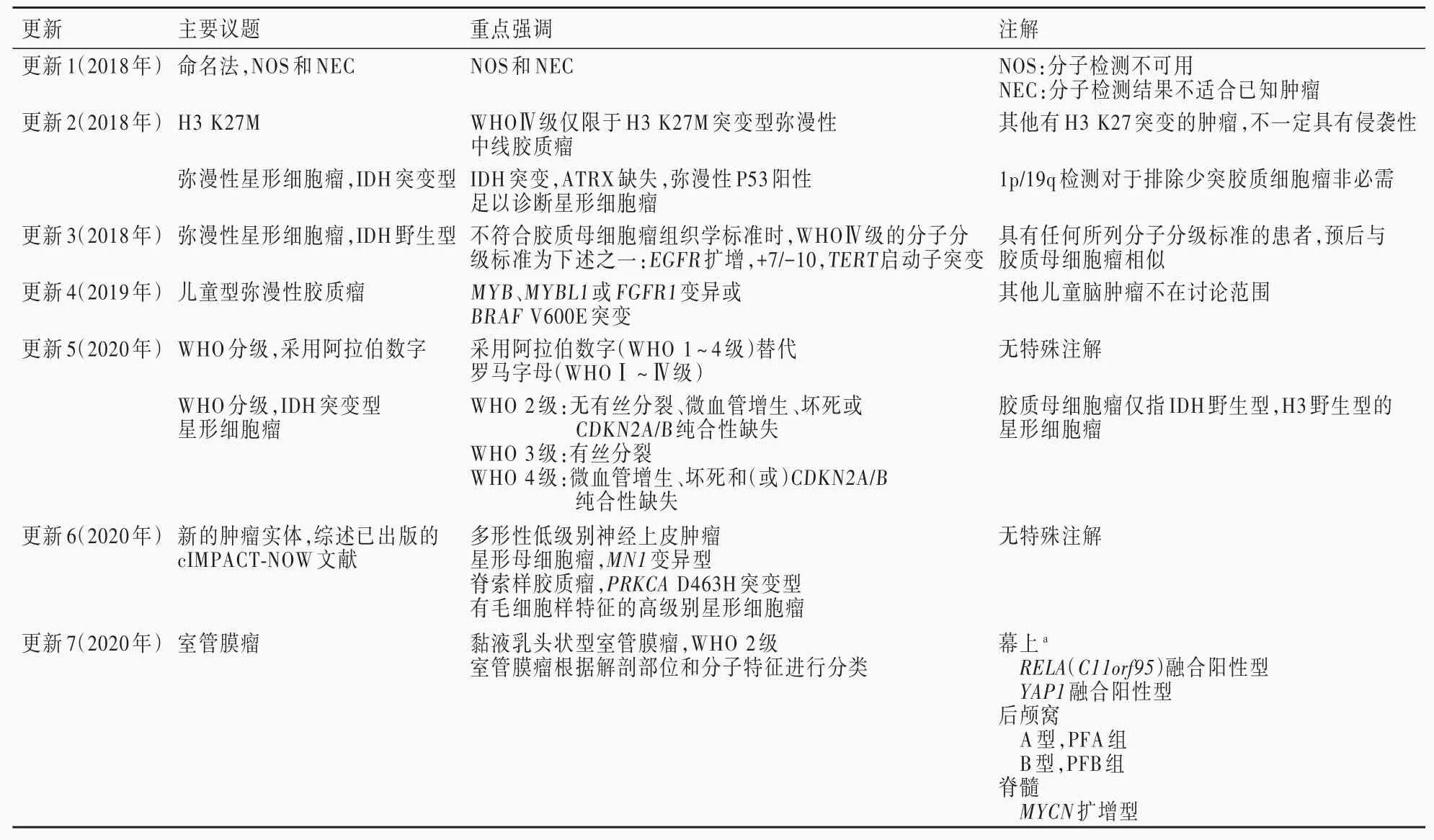
表3 cIMPACT-NOW关于中枢神经系统肿瘤分类的更新[20]Table 3. Renewal of cIMPACT-NOW on central nervous system tumor classification[20]
2021年,WHO出版新版肿瘤分类,进一步增加由生物学和分子特征定义的肿瘤新类型/亚型,最重要的变化是将儿童型弥漫性胶质瘤独立出来[21]。该版本使中枢神经系统肿瘤的诊断更加狭义和客观,有利于对生物学和基因背景类似的肿瘤实施传统或新型靶向治疗,也将促进个体化医疗、临床试验、基础研究和流行病学研究,最终改善患者生存状态[21]。新版肿瘤分类简表将不体现组织学亚型,在“蓝皮书”相应章节中再进行详述[9]。
1.胶质瘤、胶质神经元肿瘤和神经元肿瘤 包括成人型弥漫性胶质瘤、儿童型弥漫性低级别胶质瘤、儿童型弥漫性高级别胶质瘤、局限性星形细胞胶质瘤、胶质神经元和神经元肿瘤、室管膜肿瘤,新增14种肿瘤类型。成人型和儿童型弥漫性胶质瘤存在临床表型和分子特征差异。儿童型有可能发生于成人,尤其是青年,而成人型可能更少发生于儿童。成人型与儿童型的区分不仅出于概念的不同,且对调整和制定临床干预措施是十分必要的。应注意的是,发生于青年的IDH野生型弥漫性星形细胞瘤,应考虑是否为儿童型弥漫性胶质瘤所属实体[9]。(1)成人型弥漫性胶质瘤:新版肿瘤分类将成人型弥漫性胶质瘤分为星形细胞瘤,IDH突变型、少突胶质细胞瘤,IDH突变和1p/19q共缺失型、胶质母细胞瘤,IDH野生型。自第四版修订版的15种类型缩减为上述3种类型,主要是由于,对于不满足上述3个肿瘤实体诊断条件的成人型弥漫性胶质瘤,将在整合诊断中更普遍采用NOS或NEC,并在分层诊断中说明组织病理学特征和分子信息;既往组织学较难确定的肿瘤实体,如少突星形细胞瘤或弥漫性星形细胞瘤,IDH野生型,现在可通过分子特征客观判断,少突星形细胞瘤这一含混不清的实体被废除[9]。新版肿瘤分类采用肿瘤类型内分级方式,减少独立实体。①星形细胞瘤,IDH突变型以及少突胶质细胞瘤,IDH突变和1p/19q共缺失型。第一版分类将弥漫性星形细胞瘤分为纤维型、原浆型、肥胖细胞型,至第四版修订版已删除纤维型、原浆型,仅保留肥胖细胞型。根据肿瘤类型内分级原则,星形细胞瘤,IDH突变型为CNS WHO 2~4级,少突胶质细胞瘤,IDH突变和1p/19q共缺失型为CNS WHO 2~3级[4-9]。②胶质母细胞瘤,IDH野生型。第一版分类将胶质母细胞瘤列在低分化和胚胎性肿瘤下,包括有肉瘤成分的胶质母细胞瘤和巨细胞型胶质母细胞瘤2种亚型[4]。第二版分类中,胶质母细胞瘤归入星形细胞肿瘤,为恶性程度最高的星形细胞肿瘤;血管起源的肿瘤下巨怪细胞肉瘤(monstrocellular sarcoma)归入巨细胞型胶质母细胞瘤,废除巨怪细胞肉瘤类型[5]。第三版和第四版分类将胶质母细胞瘤分为原发性胶质母细胞瘤和继发性胶质母细胞瘤[6-7]。第四版修订版中,胶质母细胞瘤分为胶质母细胞瘤,IDH野生型(约占90%)和胶质母细胞瘤,IDH突变型(约占10%),分别对应原发性和继发性胶质母细胞瘤。胶质母细胞瘤,NOS专为IDH未完全检测的肿瘤保留。胶质母细胞瘤,IDH野生型包括3种亚型,即巨细胞型胶质母细胞瘤、胶质肉瘤、上皮样胶质母细胞瘤(新亚型)[8]。新版肿瘤分类仅保留胶质母细胞瘤,IDH野生型,组织学亚型将在“蓝皮书”相应章节再进行详述[9]。多形性胶质母细胞瘤传统上指经典的胶质母细胞瘤,不包括胶质母细胞瘤亚型及一些少见的组织学形式。从命名规范角度看,多形性胶质母细胞瘤并不能替代外延更广泛的胶质母细胞瘤。除WHO中枢神经系统肿瘤分类正式确认的组织学亚型,第四版及其修订版中还提及一些少见组织学形式,如横纹肌样型胶质母细胞瘤、有原始神经元成分的胶质母细胞瘤、小细胞型胶质母细胞瘤、有少突胶质细胞瘤成分的胶质母细胞瘤[7-8],以及见诸文献报道的有腺样型、上皮样型、富于脂质上皮样型、真上皮分化型、脂质化型、脂肪化生型、颗粒细胞型、有原始神经外胚层肿瘤样成分的胶质母细胞瘤[22]。多数病理学家认为,确定其为胶质母细胞瘤新亚型,不仅有异向分化的组织学表现,还需更多的临床和病理学数据,尤其需确认预后和治疗反应是否优于经典的胶质母细胞瘤[8]。WHO中枢神经系统肿瘤分类即使无法确认其为胶质母细胞瘤新亚型,这些少见的组织学形式对鉴别诊断、避免误诊和误治同样具有重要意义。例如,横纹肌样型胶质母细胞瘤可误诊为非典型性畸胎样/横纹肌样肿瘤(AT/RT)、横纹肌样型脑膜瘤以及原发性或转移性真横纹肌肿瘤;腺样型、上皮样型、富于脂质上皮样型和真上皮分化型胶质母细胞瘤易误诊为转移癌或碰撞瘤;脂质化型胶质母细胞瘤易与多形性黄色瘤型星形细胞瘤混淆;脂肪化生型胶质母细胞瘤易与脂肪神经细胞瘤和中枢神经系统原始神经外胚层肿瘤混淆;颗粒细胞型胶质母细胞瘤易与颗粒细胞瘤混淆;有原始神经外胚层肿瘤样成分的胶质母细胞瘤易与中枢神经系统原始神经外胚层肿瘤和神经母细胞瘤混淆;富于脂质上皮样型、脂质化型和颗粒细胞型胶质母细胞瘤易与组织细胞增生性病变混淆[22]。胶质母细胞瘤,IDH野生型多具有共性的核心信号转导通路[视网膜母细胞瘤基因(RB)、P53、受体酪氨酸激酶(RTK)、端粒酶逆转录酶(TERT)]基因组改变。多形性胶质母细胞瘤的生物学行为和肿瘤微环境(缺氧、免疫细胞浸润、新生血管形成等)也可影响转录谱特征。根据多形性胶质母细胞瘤基因组与转录谱的组合特征,分为3种分子亚型,即前神经型,基因表达/RTKⅠ/LGm6 DNA甲基化组,其标志是CDK4和PDGFRA扩增,在年轻成人中最常见;经典型,基因表达/RTKⅡDNA甲基化组,表现为高频EGFR扩增和CDKN2A/B纯合性缺失;间质型/间质样型,表现为NF1缺失和肿瘤内巨噬细胞浸润增加。多数情况下,上述3种分子亚型在同一多形性胶质母细胞瘤中重叠出现,均与TERT启动子突变有关。虽然多形性胶质母细胞瘤转录谱尚未被WHO采纳用于肿瘤分类,目前也无对应的治疗策略,但可加深对肿瘤异质性和进化的理解,促进靶向治疗策略的探索[23]。大脑胶质瘤病在第四版修订版中已不再作为肿瘤实体,但仍有临床医师使用这一诊断,在此有必要澄清。第一版分类将大脑胶质瘤病作为实体,归属低分化和胚胎性肿瘤;第二版分类归属来源未定的神经上皮肿瘤;第三版分类列为来源未定的胶质肿瘤;第四版分类列为星形细胞肿瘤,是指肿瘤呈弥漫浸润性生长,累及多个脑叶,也累及基底节、脑干甚至脊髓和小脑,可为星形细胞分化或者少突胶质细胞分化,也可恶性去分化为胶质母细胞瘤;第四版修订版中,广泛侵袭浸润形式可见于许多弥漫性胶质瘤,包括弥漫性星形细胞瘤,IDH突变型、少突胶质细胞瘤,IDH突变和1p/19q共缺失型和胶质母细胞瘤,IDH野生型,大脑胶质瘤病并非独立类型[8]。(2)儿童型弥漫性低级别胶质瘤:新版肿瘤分类将儿童型弥漫性低级别胶质瘤分为弥漫性星形细胞瘤,MYB或MYBL1变异型、青年人多形性低级别神经上皮肿瘤、血管中心型胶质瘤、弥漫性低级别胶质瘤,MAPK通路变异型4种类型,其共同特征是肿瘤在脑实质内呈弥漫性生长,但组织学形态存在重叠并缺乏特征性,需通过分子特征进行定性。弥漫性低级别胶质瘤,MAPK
通路变异型在组织学形态上可以是星形细胞瘤或少突胶质细胞瘤;从分子特征角度看,MAPK通路变异型包括FGFR1酪氨酸激酶结构域(TKD)重复、FGFR1突变、FGFR1融合以及BRAFV600E突变、BRAF融合、BRAF插入突变,在整合诊断和分层诊断中将综合完整的肿瘤信息。血管中心型胶质瘤已于第四版修订版中增为新类型[9,18]。(3)儿童型弥漫性高级别胶质瘤:新版肿瘤分类将儿童型弥漫性高级别胶质瘤分为弥漫性中线胶质瘤,H3 K27变异型、弥漫性半球胶质瘤,H3 G34突变型、弥漫性儿童型高级别胶质瘤,H3野生和IDH野生型、婴儿型半球胶质瘤4种类型,后3种为新增类型。弥漫性中线胶质瘤,H3 K27变异型已于第四版修订版中增为新类型,当时命名为弥漫性中线胶质瘤,H3 K27M突变型,但现在发现,除H3 K27突变(H3 K27M或H3 K27I突变)外,EGFR突变或H3野生伴EZHIP过表达也可致病。儿童型弥漫性高级别胶质瘤同样需要分子特征及整合组织病理学和分子信息综合诊断。婴儿型半球胶质瘤为新生儿期或婴儿期发病,特征性融合基因包括ALK、ROS1、NTRK1/2/3或MET。胶质母细胞瘤在儿童型肿瘤中不再作为诊断术语[9,18]。其他新增类型参见本期专题《2021年世界卫生组织中枢神经系统肿瘤分类(第五版)儿童型弥漫性胶质瘤分类解读》的相关内容[24]。(4)局限性星形细胞胶质瘤:新版肿瘤分类将其分为6种类型。“局限性”相对于“弥漫性”,此类肿瘤生长模式“坚实”,但并不意味低级别的生物学行为。第一版分类即将毛细胞型星形细胞瘤、室管膜下巨细胞型星形细胞瘤列为星形细胞肿瘤,第二版分类将多形性黄色瘤型星形细胞瘤也列为星形细胞肿瘤[4-5],毛细胞型星形细胞瘤和多形性黄色瘤型星形细胞瘤无IDH突变但常有BRAF突变或融合;室管膜下巨细胞型星形细胞瘤作为结节性硬化症的伴发肿瘤,具有TSC1/2突变[8]。毛细胞黏液型星形细胞瘤作为毛细胞型星形细胞瘤的组织学亚型纳入第四版分类,二者均可为Ⅰ型神经纤维瘤病(NF1)的中枢神经系统伴发肿瘤。当时认为,毛细胞黏液型星形细胞瘤具有侵袭性,易局部复发和脑脊液播散,其预后差于典型的毛细胞型星形细胞瘤,定义为WHOⅡ级[7]。此后发现,部分毛细胞黏液型星形细胞瘤可随时间成熟为毛细胞型星形细胞瘤,二者组织学和基因型具有广泛重叠,侵袭性生物学行为尚不确定,第四版修订版定义为WHOⅠ级[8]。有毛细胞样特征的高级别星形细胞瘤的组织学形态呈现毛细胞样特征,常见MAPK信号转导通路基因变异,如NF1突变、BRAF突变和融合、FGFR1突变和融合、KRAS突变,以及合并CDKN2A/B纯合性缺失和ATRX表达缺失,同时具有特定的DNA甲基化谱。发病部位主要为小脑,可原发亦可继发于低级别肿瘤,平均发病年龄约40岁,CNS WHO 3级(尚待临床数据进一步确定)[9,13]。脊索样胶质瘤是第三版分类增加的肿瘤实体,列于来源未定的胶质肿瘤下。当时发现的病例均发生于成人第三脑室,故命名为第三脑室脊索样胶质瘤,后来在其他部位也有发现,故新版肿瘤分类删除部位信息[6,9]。星形母细胞瘤,MN1变异型具有星形母细胞瘤的组织学形态,分子特征是MN1变异,通常为MN1-BEND2融合,多见于青年,主要位于幕上,影像学多呈边界清晰的囊性或囊实性占位,但在组织病理学和分子信息方面尚待进一步明确,以资与组织学形态和基因变异相似的其他神经上皮肿瘤相鉴别,因依据不足CNS WHO分级尚未确定[9,13]。(5)胶质神经元和神经元肿瘤:是一组具有神经元分化特征的肿瘤,包括14种类型,其中3种为新增类型。神经节细胞胶质瘤和神经节细胞瘤是熟知类型。第二版分类增加类型为小脑发育不良性神经节细胞瘤,是Cowden综合征的颅内伴发肿瘤,为PTEN胚系变异所致[5]。婴儿促纤维增生性神经节细胞胶质瘤/婴儿促纤维增生性星形细胞瘤在第三版分类中更名为婴儿促纤维增生性星形细胞瘤/神经节细胞胶质瘤;中枢神经细胞瘤是好发于侧脑室室间孔区的脑室内神经元性肿瘤;小脑脂肪神经细胞瘤为第三版分类增加类型,新版肿瘤分类仍为CNS WHO 2级,发生于成人小脑,平均发病年龄为50岁,无性别差异,组织学特征为在具有神经细胞形态特点的小肿瘤细胞的单调背景下,存在簇状排列的脂化细胞,类似于脂肪细胞[6]。第四版分类增加类型有脑室外神经细胞瘤,发生于脑室系统外的脑实质,组织学形态和生物学行为与中枢神经细胞瘤相似;乳头状胶质神经元肿瘤,WHOⅠ级(亦CNS WHO 1级),为罕见肿瘤实体,发病年龄范围广,好发于颞叶和额叶,常见症状有头痛和癫发作,CT和MRI显示肿瘤邻近皮质或脑室生长,为边界清晰的肿物,有时呈囊-壁结节形式,实性成分呈T1WI等或低信号,T2WI等或高信号,增强扫描呈强化征象;形成菊形团的胶质神经元肿瘤,当时称为第四脑室形成菊形团的胶质神经元肿瘤,WHOⅠ级(亦CNS WHO 1级),为生长缓慢的少见肿瘤,好发于青年,梗阻性脑积水和共济失调是最常见的临床表现,肿瘤常占据第四脑室和(或)中脑导水管,可向小脑延伸生长,T2WI表现为边界清晰的高信号[7]。第四版修订版增加类型为弥漫性软脑脊膜胶质神经元肿瘤,亦称为儿童播散性少突胶质细胞样软脑脊膜肿瘤,表现为弥漫性软脑脊膜病变,伴或不伴可识别的实质成分(常见于脊髓),好发于儿童和青少年,肿瘤的疾病分类学仍不清楚,有些病理学和基因特征提示与毛细胞型星形细胞瘤或胶质神经元肿瘤有关,预后不一致,有些肿瘤生长相当缓慢,但继发性脑积水致残应引起重视[8]。新版肿瘤分类新增类型包括有少突胶质细胞瘤样特征和核簇的弥漫性胶质神经元肿瘤(暂定类型)、黏液样胶质神经元肿瘤(暂定类型)、多结节和空泡状神经元肿瘤(第四版修订版已提及)[9]。新增肿瘤类型特点参见本期专题《2021年世界卫生组织中枢神经系统肿瘤分类(第五版)新增肿瘤介绍》相关解读[25]。(6)室管膜肿瘤:第一版分类中,室管膜肿瘤和脉络丛肿瘤同属一类。室管膜瘤包括3种组织学亚型,即黏液乳头状型、乳头状型和室管膜下室管膜瘤[4]。第二版分类调整组织学亚型为细胞型、乳头状型、透明细胞型,黏液乳头状型室管膜瘤和室管膜下室管膜瘤作为肿瘤实体[5]。第三版分类增加伸长细胞型室管膜瘤这一新亚型[6]。第四版修订版中,室管膜瘤的组织学亚型为乳头状型、透明细胞型、伸长细胞型,细胞型因组织学表现与典型的室管膜瘤具有广泛重叠而删除[8]。新版肿瘤分类鉴于室管膜瘤的组织学亚型对临床预后的预测无提示意义,故删除所有组织学亚型,替代为组织学形态描述[9]。第四版修订版也尝试以整合诊断的模式对室管膜瘤进行分类和分级,但由于当时在临床价值、预后意义和可重复性方面均不成熟,仅命名室管膜瘤,RELA融合阳性型[8]。新版肿瘤分类中,肿瘤实体根据组织学和分子特征以及解剖部位命名。幕上室管膜瘤分为2种分子分型,即幕上室管膜瘤,ZFTA融合阳性型和幕上室管膜瘤,YAP1融合阳性型。后颅窝室管膜瘤也分为2种分子分型,PFA组以婴幼儿为主,以H3 K27me3表达缺失、CXorf67过表达为分子特征;PFB组以大龄儿童或成人为主,H3 K27me3表达正常。以MYCN扩增为特征的脊髓室管膜瘤定义为脊髓室管膜瘤,MYCN扩增型。如果室管膜瘤的分子检测与上述分子改变均不同,或者分子检测失败或不可用时,则以解剖部位定义室管膜瘤,后缀NEC或NOS。虽然甲基化组学研究可鉴定上述肿瘤,但目前DNA甲基化状态并不能为这两种肿瘤提供额外的临床病理价值。应注意的是,黏液性乳头状型室管膜瘤由于复发可能性与传统脊髓室管膜瘤相似,目前认为是CNS WHO 2级,不再是既往分类中的WHOⅠ级[9,19]。
2.脉络丛肿瘤 由于脉络丛肿瘤具有明显的上皮细胞特征,而原发性神经上皮肿瘤更多的是向胶质和(或)神经元分化,很少上皮分化,故新版肿瘤分类将脉络丛肿瘤与原发性神经上皮肿瘤区分开来[9]。第一版分类将脉络丛肿瘤分为脉络丛乳头状瘤和间变性(恶性)脉络丛乳头状瘤[4]。第二版和第三版分类中,分为脉络丛乳头状瘤和脉络丛癌,二者有不同的发生过程。脉络丛乳头状瘤是良性肿瘤,而脉络丛癌是高度恶性肿瘤,二者之间无中间型[5-6]。第四版分类在脉络丛乳头状瘤(WHOⅠ级)与脉络丛癌(WHOⅢ级)之间增加具有中间特点的肿瘤类型,非典型性脉络丛乳头状瘤。与脉络丛乳头状瘤相比较,非典型性脉络丛乳头状瘤的有丝分裂活性增加,手术仍可治愈,但是复发的可能性增加[7]。第四版修订版和新版肿瘤分类中脉络丛肿瘤无变化[8-9]。
3.胚胎性肿瘤 新版肿瘤分类将胚胎性肿瘤分为髓母细胞瘤和其他中枢神经系统胚胎性肿瘤。原始神经外胚层肿瘤(PNET)是胚胎性肿瘤不能回避的话题。1973年,Hart和Earle发现小脑外的中枢神经系统存在组织学形态类似小脑髓母细胞瘤的肿瘤,认为其与小脑髓母细胞瘤一样,来源于神经外胚层某些不成熟的前体细胞,并首先提出“原始神经外胚层肿 瘤”的 概 念[3]。1983年,Rorke和Becker建议采用原始神经外胚层肿瘤概括不同的神经系统胚胎性肿瘤,包括髓母细胞瘤、室管膜母细胞瘤、神经母细胞瘤、松果体母细胞瘤[3]。这一建议基于这样一种假设,即所有的原始神经外胚层肿瘤均来自共同的祖细胞,在大脑半球来自室管膜下基质层的前体细胞,在小脑来自于外颗粒细胞层的前体细胞。虽然这些胚胎性肿瘤细胞自其祖细胞恶性变后,主要倾向神经细胞分化,但也具有向多种细胞谱系分化的能力,即神经细胞、星形细胞、室管膜细胞、横纹肌细胞、黑色素细胞等[3]。第二版分类为避免争议,将原始神经外胚层肿瘤作为一般术语,指小脑髓母细胞瘤以及组织学形态无法与小脑髓母细胞瘤区分的脑或脊髓肿瘤,而室管膜母细胞瘤、神经母细胞瘤、髓上皮瘤仍保留在胚胎性肿瘤类别[5]。第三版分类将幕上原始神经外胚层肿瘤(包括神经母细胞瘤和神经节细胞神经母细胞瘤亚型)与发生于小脑的髓母细胞瘤区分[6]。第四版分类重新命名为中枢神经系统原始神经外胚层肿瘤,仍指主要发生于儿童和成人的胚胎性肿瘤,具有侵袭性行为,细胞分化较差或出现向神经元、星形细胞和室管膜细胞谱系的差异分化。为将发生于脑干和脊髓的类似肿瘤包括其中,同时避免与发生于中枢神经系统以外的原始神经外胚层肿瘤相混淆,故加上中枢神经系统的前缀。除非特别指明,中枢神经系统原始神经外胚层肿瘤与幕上原始神经外胚层肿瘤是同义词,用于描述发生于小脑外的未分化或分化较差的中枢神经系统胚胎性肿瘤。如果肿瘤细胞仅向神经元分化,定义为中枢神经系统神经母细胞瘤;如果出现肿瘤性神经节细胞,称为中枢神经系统神经节神经母细胞瘤;如果出现室管膜母细胞“菊形团”结构,定义为室管膜母细胞瘤[7]。第四版修订版将中枢神经系统原始神经外胚层肿瘤从分类实体中删除,符合此诊断的实体,归属为中枢神经系统胚胎性肿瘤,NOS[8]。(1)髓母细胞瘤:最初由Bailey和Cushing根据胚胎细胞残留学说命名,尽管发育研究尚无髓母细胞瘤的证据,但出于历史沿袭,仍保留此命名[3]。髓母细胞瘤的组织学亚型为,第一版分类分为促纤维增生型和髓母肌母细胞瘤2种亚型[4];第二版分类增加黑色素型髓母细胞瘤亚型[5];第三版分类增加大细胞型髓母细胞瘤亚型[6];第四版分类认为,髓母肌母细胞瘤与黑色素型髓母细胞瘤是分化差异造成的组织学形态不均一表现,无独特的临床和遗传学特征,故不再作为独立的病理亚型,可相应描述为髓母细胞瘤伴肌原性分化和髓母细胞瘤伴黑色素细胞分化[7];第四版修订版调整髓母细胞瘤组织学亚型为经典型、促纤维增生/结节型、广泛结节型、大细胞型/间变性髓母细胞瘤4种,相对而言,促纤维增生/结节型、广泛结节型预后较好,大细胞型/间变性预后较差[8];新版肿瘤分类保持组织学亚型不变,在简表中合并为髓母细胞瘤组织学亚型条目,并在“蓝皮书”的相应章节再进行详述[9]。髓母细胞瘤的分子分型为,第四版修订版尝试将其分为WNT活化型、SHH活化型、3组型、4组型。基因表达谱比较分析显示,3组型和4组型的分子特征有很大重叠,可以合并为非WNT/非SHH活化型。SHH活化型是否存在TP53突变,在临床和病理学特征上存在显著差异[8]。新版肿瘤分类将髓母细胞瘤分子分型调整为WNT活化型、SHH活化和TP53野生型、SHH活化和TP53突变型、非WNT/非SHH活化型(包括3组型和4组型)4种亚型。通过大规模DNA甲基化谱和转录谱分析,4种主要分子亚型还可以更细致地分出新亚组。SHH活化型分为4个亚组,非WNT/非SHH活化型分为8个亚组。与髓母细胞瘤的4种主要分子亚型一样,有些亚组也与临床病理学和基因特征有关,可以为临床诊断、预后预测提供信息[9]。髓母细胞瘤的不同组织学形态模式有其特定的临床关联,分子特征定义的髓母细胞瘤与组织学形态模式有明显的联系。例如,促纤维增生/结节型、广泛结节型髓母细胞瘤均属SHH活化型,多为SHH活化型1亚组或2亚组;几乎所有的WNT活化型均具有经典的髓母细胞瘤组织学形态;大多数大细胞型/间变性髓母细胞瘤属SHH活化型3亚组或3组型/4组型2亚组[9]。第四版修订版和新版肿瘤分类均以矩阵模块方式,分列出组织学确定的亚型和分子特征确定的亚型,按照整合和分层原则进行诊断,在适当情况下,也有NOS和NEC做后缀选项[8-9]。(2)其他中枢神经系统胚胎性肿瘤:除髓母细胞瘤外的胚胎性肿瘤,包括非典型性畸胎样/横纹肌样肿瘤、有多层菊形团的胚胎性肿瘤、筛状神经上皮肿瘤(暂定类型)、中枢神经系统神经母细胞瘤,FOXR2活化型、有BCOR内部串联重复的中枢神经系统肿瘤和中枢神经系统胚胎性肿瘤,其中第四版分类已纳入非典型性畸胎样/横纹肌样肿瘤[6],第四版修订版已纳入有多层菊形团的胚胎性肿瘤[8]。新版肿瘤分类进一步确认非典型性畸胎样/横纹肌样肿瘤的3种分子亚型;有多层菊形团的胚胎性肿瘤,除较常见的C19MC变异型,新增DICER1突变型。应注意的是,有少部分非典型性畸胎样/横纹肌样肿瘤伴发肾脏横纹肌样肿瘤,而DICER1胚系变异亦可导致常染色体显性遗传性疾病。虽然新版肿瘤分类将有BCOR内部串联重复的中枢神经系统肿瘤作为新实体,但并未肯定其为神经外胚层来源,这是由于一些组织学形态相似的肉瘤也有BCOR外显子15内部串联重复的报告,目前这些肿瘤是神经上皮肿瘤还是间质肿瘤尚无共识,其疾病分类可以根据未来发现而相应调整。筛状神经上皮肿瘤是新版肿瘤分类新增的暂定类型。此外,凡不符合特定诊断、需以NEC或NOS后缀的胚胎性肿瘤均归为中枢神经系统胚胎性肿瘤,组织学形态和分子特征可以通过整合及分层诊断报告体现,以便清晰、有效地反映相关肿瘤的个性特征[9]。
4.松果体肿瘤 第一版分类仅列出松果体细胞肿瘤,包括松果体细胞瘤和松果体母细胞瘤[4];第二版分类更名为松果体实质性肿瘤,增加混合性/过渡性松果体肿瘤;在第三版分类中被中分化的松果体实质性肿瘤替代[5-6];第四版分类改为松果体区肿瘤,包括松果体细胞瘤、中分化的松果体实质性肿瘤、松果体母细胞瘤、松果体区乳头状肿瘤(新增实体),由于松果体区乳头状肿瘤的组织学形态不同于松果体实质性肿瘤,改为松果体区肿瘤[7];第四版修订版无变化[8];新版肿瘤分类改为松果体肿瘤,原有实体不变,新增松果体区促纤维增生性黏液样肿瘤,SMARCB1突变型,是一种缺乏恶性组织病理学征象的罕见SMARCB1突变肿瘤[9]。中分化的松果体实质性肿瘤、松果体区乳头状肿瘤、松果体区促纤维增生性黏液样肿瘤,SMARCB1突变型这3个实体的生物学行为和组织学分级标准仍有诸多待定问题。分子特征也在松果体肿瘤的诊断中有重要体现,如KBTBD4框内插入可作为中分化的松果体实质性肿瘤的诊断标准。通过DNA甲基化谱,松果体母细胞瘤分为4种分子亚型,即松果体母细胞瘤,微小RNA(miRNA)加工变异1型,见于儿童,其特征是DICER1、DROSHA或DGCR8突变;松果体母细胞瘤,miRNA加工变异2型,多见于大龄儿童,预后相对较好,其特征是DICER1、DROSHA或DGCR8突变;松果体母细胞瘤,MYC/FOXR2活化型,见于婴儿,有MYC活化和FOXR2过表达;松果体母细胞瘤,RB1突变型,见于婴儿,有RB1胚系变异,双眼视网膜母细胞瘤与松果体母细胞瘤可能同时发病,此时称为三侧性视网膜母细胞瘤[9]。
5.颅神经和椎旁神经肿瘤 第一版分类仅列出神经鞘细胞肿瘤,包括神经鞘瘤、间变性(恶性)神经鞘瘤、神经纤维瘤、间变性(恶性)神经纤维瘤[4];第二版分类改为颅神经和脊神经的肿瘤;第三版分类又改为周围神经肿瘤[5-6];第四版及其修订版和新版肿瘤分类均确定为颅神经和椎旁神经肿瘤,包括神经鞘瘤、神经纤维瘤、神经束膜瘤、杂合性神经鞘膜瘤、恶性黑色素性神经鞘膜瘤、恶性周围神经鞘膜瘤、副神经节瘤,各亚型在“蓝皮书”的相应章节中再进行详述[7-9]。新版肿瘤分类中,副神经节瘤涉及交感神经和副交感神经系统中特化的神经内分泌细胞,现归为神经肿瘤[9]。由于免疫组化和DNA甲基化特征有别于其他部位常见副神经节瘤,且缺乏家族关联性,马尾/终丝区副神经节瘤被认为是一种独特的肿瘤类型。第二版分类在神经鞘瘤下增加黑色素亚型,并在第四版修订版中成为独立实体,命名为黑色素性神经鞘瘤[5,8]。新版肿瘤分类认为这是一种特殊的肿瘤类型,具有独特的分子特征,常具有侵袭性,与其他神经鞘膜肿瘤包括神经鞘瘤不同,参照WHO软组织肿瘤分类,将其更名为恶性黑色素性神经鞘膜瘤。新版肿瘤分类的神经纤维瘤也增加一个新的组织学亚型,即生物学潜能未知的非典型性神经纤维瘤性肿瘤(atypical neurofibromatous neuoplasm),是Ⅰ型神经纤维瘤病的相关肿瘤,具有恶性转化特征,但尚不足以明确诊断为恶性周围神经鞘膜肿瘤[9]。
6.脑膜瘤 第一版分类在脑膜瘤下进行组织学亚型的分型,但是存在错误,例如将血管外皮细胞瘤作为脑膜瘤的亚型[4];第二版分类进行调整,并增加新亚型,但也存在实体与亚型不分的问题,如将乳头状型脑膜瘤、非典型脑膜瘤和间变性脑膜瘤作为实体[5];第三版分类将脑膜瘤分为15种亚型,WHOⅠ级的脑膜瘤亚型为脑膜皮型、纤维型、过渡型、砂粒体型、血管瘤型、微囊型、分泌型、富于淋巴细胞-浆细胞型、化生型;WHOⅡ级的脑膜瘤亚型为非典型性、透明细胞型、脊索样型;WHOⅢ级的脑膜瘤亚型为乳头状型、横纹肌样型、间变性[6];第四版及其修订版无变化[7-8]。新版肿瘤分类中,脑膜瘤仍认为是单一的实体,15种亚型反映出其组织学形态的广泛性。该版本强调,确定非典型性(CNS WHO 2级)或间变性(CNS WHO 3级)脑膜瘤的分级标准适用于任何亚型。第一版至第四版修订版认为,脊索样型和透明细胞型脑膜瘤的复发率高于前述WHOⅠ级脑膜瘤亚型,故直接定义为WHOⅡ级。目前认为,CNS WHO 2级的脑膜瘤亚型应通过更大规模的前瞻性研究验证并纳入,寻找附加的预后生物学标志物。按照第一版至第四版修订版标准,横纹肌样型和乳头状型无需考虑任何其他恶性指标,仅组织学形态即符合WHOⅢ级。而乳头状型和横纹肌样型组织学特征常与其他侵袭性特征组合出现,提示其分级不应仅依靠横纹肌样细胞或乳头状结构。一些分子生物学标志物也与脑膜瘤分类和分级有关,包括SMARCE1突变(透明细胞型)、BAP1突变(横纹肌样型和乳头状型)、KLF4/TRAF7突变(分泌型)、TERT启动子突变和(或)CDKN2A/B纯合性缺失(CNS WHO 3级),H3 K27me3胞核表达缺失(预后更差可能)和DNA甲基化谱(预后分型)[9]。
7.间叶性非脑膜皮肿瘤 分为软组织肿瘤、软骨及骨肿瘤、脊索肿瘤3大类。新版肿瘤分类中,力求间叶性非脑膜皮肿瘤的相关术语与WHO骨和软组织肿瘤“蓝皮书”保持一致,目前仅包括只发生于中枢神经系统的肿瘤或者与对应软组织肿瘤相似但常发生于中枢神经系统的肿瘤。一些常见的软组织肿瘤(如平滑肌瘤)罕见于中枢神经系统,由于其诊断特征与对应的软组织肿瘤相同,不包括在此类别下。新增类型有颅内间质性肿瘤,FET-CREB融合阳性型(暂定类型)、CIC重排肉瘤、原发性颅内肉瘤,DICER1突变型[9]。2016年以前,血管外皮细胞瘤这一命名主要由神经病理学家使用,软组织病理学家称为孤立性纤维性肿瘤。第四版修订版创造一联合术语——孤立性纤维性肿瘤/血管外皮细胞瘤,用以描述这一高复发率和具有颅外转移风险的病变[8]。突破既往中枢神经系统肿瘤中一个类型/亚型仅对应一个特定WHO分级的传统,在孤立性纤维性肿瘤/血管外皮细胞瘤同一实体中,设3个WHO分级,即按照传统的以罗马数字标识的WHOⅠ~Ⅲ级,同时也参照非中枢神经系统肿瘤,以阿拉伯数字标识的CNS WHO 1~3级。新版肿瘤分类删除血管外皮细胞瘤,直接称为孤立性纤维性肿瘤,与软组织肿瘤的病理诊断术语保持一致,但孤立性纤维性肿瘤的WHO分级方法还有其发病部位的特殊性,与其他中枢神经系统以外的孤立性纤维性肿瘤有所区别[9]。
8.黑色素细胞肿瘤 新版肿瘤分类按照弥漫性脑膜黑色素细胞肿瘤和局限性脑膜黑色素细胞肿瘤进行亚分类,与第四版修订版相比较,肿瘤实体无变化[8-9]。
9.血液和淋巴肿瘤与组织细胞肿瘤 淋巴瘤和组织细胞肿瘤在过去10余年已发生很多变化。第四版修订版已将发生于中枢神经系统的血液和淋巴肿瘤与组织细胞肿瘤实体进一步扩展,并与全身性造血/淋巴组织肿瘤的相应类别平行一致[8]。新版肿瘤分类仅纳入中枢神经系统出现相对较多、具有特殊组织学或分子特征的淋巴瘤或组织细胞肿瘤实体[9]。此类疾病的完整谱系参阅相应的WHO造血及淋巴系统肿瘤分类。
10.生殖细胞肿瘤 新版肿瘤分类无变化[9]。
11.鞍区肿瘤 牙釉质细胞瘤型颅咽管瘤和乳头状型颅咽管瘤在第一版至第四版修订版中均为颅咽管瘤的2种亚型[4-8]。二者在发病年龄,影像学、组织病理学和分子特征,DNA甲基化谱方面均存在不同,新版肿瘤分类将其作为两种不同的肿瘤类型[9]。新版肿瘤分类的另一变化为,将垂体细胞瘤,鞍区颗粒细胞瘤和梭形细胞嗜酸细胞瘤划归在一起,作为相关肿瘤类型组[9]。尽管上述种类可能仅代表相同肿瘤的组织学形态变化,但其发病年龄和临床结局并不相同,因此仍认为其是不同的实体。垂体腺瘤由于已在WHO内分泌肿瘤分类中叙述,第三版至第四版修订版的鞍区肿瘤下均删除这一实体[6-8]。新版肿瘤分类中,垂体腺瘤重归鞍区肿瘤类别,并沿用第四版WHO内分泌肿瘤分类,按照垂体激素免疫组化染色并结合转录因子,遵循腺垂体的细胞谱系划分[9]。新版肿瘤分类还采用垂体神经内分泌肿瘤(PitNET)这一新术语,由WHO内分泌组提出,并将在第五版WHO内分泌肿瘤分类中进一步讨论。新版肿瘤分类还新增一个实体——垂体母细胞瘤,为少见的婴儿期胚胎性肿瘤,系由原始原胚细胞、神经内分泌细胞、Rathke上皮细胞组成[9]。
12.中枢神经系统转移性肿瘤 分为脑和脊髓实质的转移性肿瘤和脑膜的转移性肿瘤。随着中枢神经系统外一些特定肿瘤治疗的进步,期待免疫组化和分子诊断标志物有助于指导中枢神经系统转移性肿瘤的诊断与治疗[9]。
三、中枢神经系统肿瘤分级
任何肿瘤的分级系统应满足两个基本要求:肿瘤级别可代表其生物学行为并预测预后;分级标准应力求客观,在不同观察者之间具有最大的可重复性。早在1926年,Bailey和Cushing即将星形细胞瘤描述为三级,即星形细胞瘤、星形母细胞瘤和胶质(海绵)母细胞瘤[3]。历史上对中枢神经系统肿瘤比较有代表性的分级系统有Kernohan分级(1949年)、Ringertz分级(1951年)、第一版分类(1979年)、St.Anne/Mayo分级(1988年)、第二版分类(1993年),及基本沿用第二版分类分级标准并尝试按照ICD-O编码对神经系统肿瘤分级的第三版和第四版分类[1-3]。Kernohan对胶质瘤分级采用四级系统(1~4级),主要根据细胞间变百分比,即肿瘤细胞占肿瘤组织<25%为1级、25%~49%为2级、50%~75%为3级、>75%为4级。该系统还认为,低级别星形细胞瘤中如果出现高度恶性、具有胶质母细胞瘤特点的肿瘤灶,也意味着预后不良。Kernohan分级的缺点为,将纤维型星形细胞瘤和毛细胞型星形细胞瘤混为一谈;胶质母细胞瘤既可以是3级,也可以是WHO 4级;肿瘤恶性程度易错估。按照Kernohan分级,早期文献报道2级与3级星形细胞瘤无明显的临床预后差异,如果按照Ringertz三级系统,患者则具有明显的预后差异,自此三级系统开始较广泛的临床应用[1-3]。Ringertz分级及以后的修正分级法通过肿瘤名称即可确定肿瘤病理级别,如星形细胞瘤(1级)代表分化良好、有丝分裂少见、无血管增殖;间变性星形细胞瘤(2级)代表富于细胞、有丝分裂活跃、血管增殖较少、无坏死;胶质母细胞瘤(3级)代表细胞具有多形性、细胞丰富、有丝分裂多、血管增殖明显、存在坏死。毛细胞型及其他特殊类型的星形细胞瘤未被分级。与Kernohan分级相比,Ringertz分级与预后的关系更密切,但是在分级标准的客观性和可重复性上仍有缺陷[1-3]。St.Anne/Mayo分级由Daumas-Duport等于1988年在口唇鳞状细胞癌分类的基础上提出,并根据下述4种组织学特点分级,包括胞核的非典型性、有丝分裂、血管内皮细胞增殖、坏死。胞核的非典型性系指胞核形状、大小不同,伴染色质浓集;有丝分裂必须真正存在,但对有丝分裂的数目和形态无特殊要求;血管内皮细胞增殖严格限定为出现多层血管内皮细胞,并非单纯指血管密度增高或者呈肾小球样毛细血管表现而血管内皮细胞仍为单层;坏死必须是确实存在的,然而不要求一定呈假“栅栏”样,也不包括坏死早期表现。每项标准代表1分,符合0项标准为1级、1项标准为2级、2项标准为3级、3或4项标准为4级。St.Anne/Mayo分级可重复率高达94%。该系统虽为四级系统,但是由于符合1级的肿瘤非常少见(<0.25%),其他2~4级肿瘤的生存曲线明显不同,因此有学者将这一系统认为是三级系统。类似的分级还有1989年Davis提出的UCSF(University of California at San Francisco)系统[1-3]。第一版至第四版修订版对中枢神经系统肿瘤的生物学行为和肿瘤恶性程度均采用WHOⅠ~Ⅳ级表示[4-8],其中注明的分级方案是对中枢神经系统肿瘤恶性程度进行泛泛的分级,并非严格的组织学分级系统。WHOⅠ级为增殖活性低,手术可治愈的肿瘤;WHOⅡ级为浸润性肿瘤,增殖活性虽低,但常复发,并具有进展为更高级别恶性肿瘤的倾向;WHOⅢ级肿瘤具有恶性肿瘤的组织学证据,包括胞核间变、有丝分裂活跃,多数患者需接受辅助性放疗和(或)化疗;WHOⅣ级肿瘤具有恶性细胞学表现、有丝分裂活跃、坏死倾向,术前和术后进展迅速,病死性临床结局。向周围组织广泛浸润以及脑和脊髓播散是一些WHOⅣ级肿瘤的特点。WHO分级作为预测治疗反应和临床结局的标准之一,还应综合参考临床特点(如年龄和神经功能)、肿瘤部位、影像学特点(如有无强化征象)、手术切除程度、增殖指数、遗传学改变。一般说来,WHOⅡ级肿瘤患者生存期>5年,WHOⅢ级为2~3年,WHOⅣ级取决于是否接受有效治疗。多数胶质母细胞瘤患者尤其是老年患者,生存期<1年。除胶质母细胞瘤外的WHOⅣ级肿瘤,临床结局可能稍好,例如,同样为WHOⅣ级的髓母细胞瘤和生殖细胞瘤,如果未经治疗可迅速致命,但随着放疗和化疗技术的进步,二者的5年生存率分别>60%和80%。新版肿瘤分类对中枢神经系统肿瘤的分级进行较大调整,这些调整力求为下述问题提出解决方案[9]:(1)第一版至第四版修订版采用WHOⅠ~Ⅳ级表示中枢神经系统肿瘤的生物学行为和肿瘤恶性程度,但是罗马数字存在误写(打印)、误认的可能、从而影响临床诊治;而非中枢神经系统肿瘤一直采用阿拉伯数字。(2)WHO中枢神经系统肿瘤分级把不同类别的肿瘤实体通过级别比较,理想化地与临床生物学行为相关联。无论何种类别的肿瘤实体,WHOⅠ级为良性,通过手术切除可治愈;WHOⅣ级则为高度恶性,在缺乏有效治疗的情况下,相对较短时间内即死亡。相同WHO分级肿瘤的生存期大致相同,然而实际上不同类别和实体,即使WHO分级相同,临床过程和生物学行为可能有很大不同。例如,间变性星形细胞瘤与间变性脑膜瘤分属不同肿瘤类别,但均为WHOⅢ级。(3)由于中枢神经系统肿瘤实体的特异性,其临床肿瘤分级方法一直不同于其他非中枢神经系统肿瘤。后者多采用肿瘤类型内分级,如乳腺癌或前列腺癌均根据各自特定的分级系统进行分级。而WHO中枢神经系统肿瘤分级为每个实体均分配对应的级别,如弥漫性星形细胞瘤、间变性星形细胞瘤、胶质母细胞瘤,即使不标注分级,实体名称分别对应WHOⅡ级、Ⅲ级和Ⅳ级。如果以这种方式发展,在整合诊断的今天,随着肿瘤实体的增加,会走向繁杂。(4)最初的肿瘤预后相关性研究是基于自然病程,当时有效治疗方法很少。目前评估肿瘤患者的自然病程几乎不可能,因为绝大多数患者均接受治疗,从而影响总体生存率。在现代治疗方法可显著影响中枢神经系统肿瘤患者生存期的背景下,如何对肿瘤实体分级、如何指导临床,以及分级是否还有必要,均是值得商榷的问题[9]。
新版肿瘤分类分级的主要变化为:(1)与非中枢神经系统肿瘤分级保持一致,以阿拉伯数字表示,并采用肿瘤类型内分级。第四版修订版曾尝试对孤立性纤维性肿瘤/血管外皮细胞瘤进行肿瘤类型内分级,分为WHO 1~3级共3个级别[8]。肿瘤类型内分级可以提供更大的灵活性,强调肿瘤类型内的生物学相似性,而非临床行为的相似,同时也与非中枢神经系统肿瘤的分级方式保持一致。但新版肿瘤分类总体保留既往各版对肿瘤实体的分级范围,如IDH突变型星形细胞瘤仅为CNS WHO 2~4级,脑膜瘤仅为CNSWHO 1~3级。(2)传统上,中枢神经系统肿瘤分级仅基于组织学特征,但现在一些分子生物学标志物可以提供可靠的预后信息。因此,分子信息应用于肿瘤分级和预后预测。即使组织学未见血管增生和(或)坏死,对于IDH突变型星形细胞瘤,只要出现CDKN2A/B纯合性缺失,即定为CNS WHO 4级[16];同样,对于IDH野生型弥漫性星形细胞瘤,只要出现TERT启动子突变、EGFR扩增、第7号染色体获得/第10号染色体缺失的任一项或组合,也定为CNS WHO 4级[15]。第四版分类新增的肿瘤实体或亚型,由于病例数有限,分级仍是初步的或尚未分级,有待资料的补充和长期的随访。尽管如此,由于中枢神经系统肿瘤分级仍不同于非中枢神经系统肿瘤分级系统,新版肿瘤分类建议在标注肿瘤级别时采用“CNS WHO级别”一词[9]。
中枢神经系统肿瘤根据自然病程进行分级,即使某些恶性肿瘤(如髓母细胞瘤、生殖细胞瘤)经过有效治疗具有良好预后,也仍是CNS WHO 4级。因此,进行WHO中枢神经系统肿瘤分级时必须清楚这一点,才不会让肿瘤级别干扰临床决策。例如,WNT活化型髓母细胞瘤具有很强的侵袭性,如不治疗预后极差。但是基于目前有效的治疗方案,几乎所有此类患者均有较长的生存期。如果我们不了解WHO中枢神经系统肿瘤分级原则,也不追踪学习新型治疗方法,把髓母细胞瘤,WNT活化型,CNS WHO 4级等同于其他无法治疗、预后欠佳的儿童脑肿瘤,有可能因对预后的错误判断而影响临床治疗决策。相反,若因为经治疗的WNT活化型髓母细胞瘤结局良好而认为其应为CNS WHO 1级,相当于单纯手术结局良好肿瘤,也是对其真实生物学行为的误判[21]。
四、新版肿瘤分类和分级的临床实践意义
各版WHO中枢神经系统肿瘤分类均反映出某个时期和部分专家对肿瘤信息的理解状态,因此,WHO中枢神经系统肿瘤分类永远在路上,没有终极版。新版肿瘤分类同样是中枢神经系统肿瘤分类演变的一个阶段。与既往各版本相比,新版肿瘤分类引入更多的新知识,但仍持审慎推进的态度,包括类型/亚型的增加或废弃,分类学结构的调整。无论是对于全球神经系统肿瘤基础与临床研究人员、临床医师,还是对于中枢神经系统肿瘤患者,均期待可以从新版肿瘤分类和分级的进步中获益。
新版肿瘤分类中,最重要的变化是将主要发生于成人的胶质瘤与主要发生于儿童的胶质瘤进行区分。在成人型弥漫性胶质瘤中,胶质母细胞瘤仅限于IDH野生型肿瘤[21]。因此,IDH是野生型还是突变型,应及时正确检测,对临床诊治十分重要。新版肿瘤分类中依靠附加分子特征进行诊断的肿瘤类型/亚型明显增加,意味临床对分子检测提出更多和更高要求,尤其是有治疗对应的分子亚型。让卫生健康管理部门、社会保障和健康保险知晓这种变化并给予政策支持也是临床工作者的责任。
目前,中枢神经系统肿瘤的靶向治疗研究遇到瓶颈[26]。从某种程度上说,没有生物学角度的区分,不可能从治疗角度做出有成效的努力。新版肿瘤分类为下一步临床试验提出新方向。儿童型弥漫性胶质瘤基于不同组织学形态和分子特征进行分类/分型,具有重要的治疗实践意义,尤其是有些靶点已经有可用的对应药物,靶向治疗还可推延低龄儿童的放疗,但开展严谨的临床试验十分重要。儿童型肿瘤治疗中还应关注关键分子通路在正常发育中的作用,避免靶向治疗对儿童发育的潜在危害[21]。新版肿瘤分类中,胶质神经元肿瘤和神经元肿瘤以及室管膜瘤在组织学和分子特征整合诊断方面也有新的进展,下一步尚待研究如何将新认识转化为更有效的治疗。髓母细胞瘤分子分型有由亚型向亚组进一步细分的趋势,但也增加根据分子特征开展临床试验的难度。把这些碎片化的独立亚组集合起来,临床试验需在入组肿瘤的均一性和入组患者病例数方面做出平衡,否则可能影响临床试验的效能[9,21]。
WHO中枢神经系统肿瘤组织学分类已历经40余年历史,深入理解肿瘤分类原则和依据,了解分类框架、类型/亚型和分级演变,追踪和捕捉最新变化,对神经肿瘤相关领域的医师来说非常重要。WHO中枢神经系统肿瘤分类不仅可以指导临床医师提高诊疗水平,也为神经肿瘤的临床与基础研究引领方向。
利益冲突 无
猜你喜欢
传染病信息(2022年2期)2022-07-15
现代消化及介入诊疗(2021年9期)2021-11-23
昆明医科大学学报(2021年3期)2021-07-22
神经损伤与功能重建(2020年11期)2020-12-01
环球时报(2018-11-09)2018-11-09
初中生世界·七年级(2018年7期)2018-09-07
校园英语·上旬(2016年5期)2016-05-14
上海医药(2016年6期)2016-04-28
企业导报(2015年16期)2015-12-14
中国青年(1990年12期)1990-08-28
