
2021年世界卫生组织中枢神经系统肿瘤分类(第五版)新增肿瘤介绍
2021-02-10 06:20:56李智
中国现代神经疾病杂志 2021年9期
李智
2021年世界卫生组织(WHO)中枢神经系统(CNS)肿瘤分类(第五版,以下简称新版肿瘤分类)即将出版。2021年6月底先期出版的《2021年世界卫生组织中枢神经系统肿瘤分类(第五版)评述》[1]指出,自2016年WHO中枢神经系统肿瘤分类第四版修订版(以下简称第四版修订版)出版及2018年1月至2020年6月中枢神经系统肿瘤分子信息与分类实践联盟-非WHO官方组织(cIMPACT-NOW)共7次更新以来,对中枢神经系统肿瘤在组织学形态、分子遗传学变异和生物学行为及其相关机制方面又有了较多新的认识,发现了一些新的肿瘤类型和亚型(表1)。新版肿瘤分类新增22个肿瘤,有些是原肿瘤类型中具有特殊分子遗传学变异的新亚型(如室管膜肿瘤中后颅窝室管膜瘤,PFA组和PFB组),其自身并不具有独特的组织学形态特征;有些是新定义的具有特殊组织学形态和分子遗传学特征的新病种(如多结节和空泡状神经元肿瘤等);还有些是尚未能确定分化的软组织肿瘤(如CIC重排肉瘤等)。尽管分子遗传学变异是新版肿瘤分类的重要原则,对肿瘤发生机制、治疗和预后均有重要指导意义,但仍强调组织学形态是肿瘤诊断的重点。本文拟对新版肿瘤分类中具有组织学形态特征的新增肿瘤进行简要介绍,以期了解这些新增肿瘤的特点。

表1 2021年WHO中枢神经系统肿瘤分类(第五版)新增肿瘤类型Table 1. Newly identified tumor types in the 2021 WHO Classification of Tumors of the Central Nervous System(fifth edition)
一、多结节和空泡状神经元肿瘤
多结节和空泡状神经元肿瘤(MVNT)是新定义的肿瘤类型,由大小不等、散在分布或聚集成团的结节构成,结节内可见形态单一的具有神经元特征的肿瘤细胞,核周和肿瘤间质内可见大小不等的空泡。多结节和空泡状神经元肿瘤最早于2013年由Huse等[2]提出,认为是一种发生于成人的癫相关良性病变。第四版修订版将其作为神经节细胞瘤的特殊组织学形态介绍,2020年cIMPACT-NOW更新6将其拟定为独立的肿瘤类型,并在新版肿瘤分类中作为胶质神经元和神经元肿瘤的一种独立类型,相当于CNS WHO 1级。
多结节和空泡状神经元肿瘤好发于成人(平均发病年龄40岁)幕上颞叶和额叶,多无临床症状或仅有癫发作和非局限性头痛,病变位于正常皮质内侧面,常围绕在脑沟周围,主要是深部皮质下带与邻近浅表皮质下白质之间。结节通常呈细小分散的圆形或卵圆形,边缘清晰,结节间为正常脑实质与星形胶质细胞分隔。影像学无占位效应和瘤周水肿,病灶不强化或仅有边缘点状强化(图1)。组织学形态,结节内肿瘤细胞为体积中等或偏大的神经元样细胞,散乱分布或沿薄壁血管分布,部分伴少突胶质细胞样胞质空亮的细胞成分,但缺乏神经节细胞瘤常见的胞体大、胞核大、核仁明显的发育异常神经元和嗜酸性颗粒小体及炎性细胞浸润,核分裂象未见(图2a,2b)。免疫组化染色,神经元样肿瘤细胞不表达胶质纤维酸性蛋白(GFAP),较一致表达HuC/HuD神经元相关抗原和少突胶质细胞转录因子2(Olig-2),胞质突触素(Syn)呈弱阳性。此外,神经元成分表达非磷酸化的相对分子质量为200×103的神经微丝蛋白(NF),而不表达神经元核抗原(NeuN)、嗜铬素A(CgA)和磷酸化NF(图2c,2d)。这些神经元成分自身并不表达CD34,但在病灶邻近区域可见CD34呈树突状阳性的神经细胞。Ki-67抗原标记指数≤1%,且Ki-67阳性细胞是结节内非神经元成分。迄今并未发现多结节和空泡状神经元肿瘤存在KIAA1549-BRAF融合、BRAFV600E突变以及IDH1/2、ATRX、TP53、TERT、CIC、FUBP1、PRKCA、CDKN2A和FGFR1基因变异,但存在丝裂原激活蛋白激酶(MAPK)信号转导通路活化基因变异(包括MAP2K1外显子2突变,非V600E位点的BRAF基因突变与FGFR2基因融合),上述基因变异提示肿瘤性病变而非发育畸型或错构瘤。多结节和空泡状神经元肿瘤多为偶然发现,如无明显临床症状,可不予处理,通常不危及患者生命。
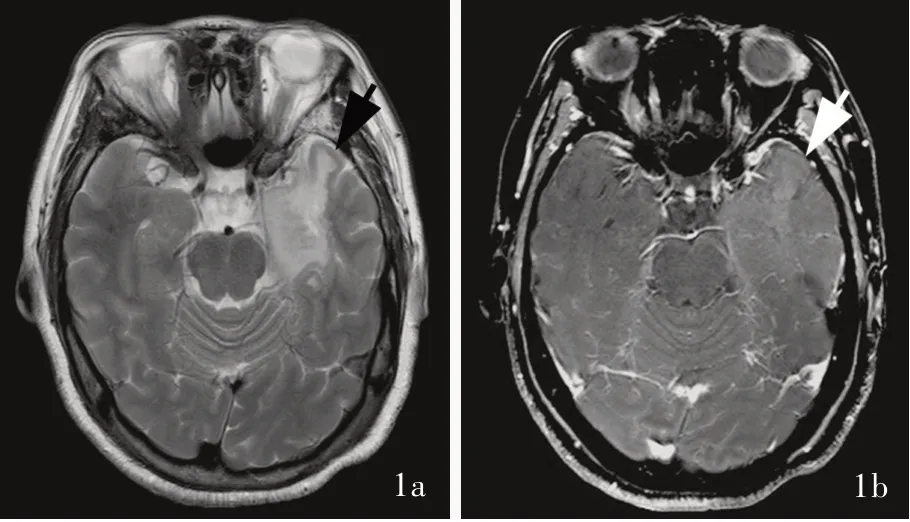
图1 多结节和空泡状神经元肿瘤的影像学特征 1a 横断面T2WI显示,左侧颞叶肿胀,伴异常高信号,部分病变位于皮质下白质浅层(箭头所示) 1b 横断面增强抑脂T1WI显示,病变区未见明显异常强化(箭头所示)Figure 1 Radiological features of MVNT Axial T2WI showed edema of left temporal lobe with a mild hyperintense lesion.Some lesion was located in the super ficial layer of subcortical white matter(arrow indicates,Panel 1a). Axial enhanced fat suppression T1WI showed no enhancement after gadolinium administration(arrow indicates,Panel 1b).
二、黏液样胶质神经元肿瘤
黏液样胶质神经元肿瘤(MGT)系一种新定义的界限清晰的胶质神经元肿瘤,通常位于隔区(隔核,有时位于透明隔),偶见于脑室周围白质或胼胝体。组织学形态为少突胶质细胞样肿瘤细胞分布于黏液样/富含黏蛋白的间质中,有时可见“漂浮”的神经元、神经细胞“菊形团”和(或)围血管神经毡结构。临床进程缓慢,相当于CNS WHO 1级。
黏液样胶质神经元肿瘤由于具有丰富的黏液样间质和少突胶质细胞样细胞特点,既往曾描述为透明隔胚胎发育不良性神经上皮肿瘤(DNT)样肿瘤[3]或脑室内胚胎发育不良性神经上皮肿瘤[4]。与胚胎发育不良性神经上皮肿瘤不同,黏液样胶质神经元肿瘤缺乏皮质内界限清晰的多结节状黏液性结节,亦无BRAF和FGFR1突变或重排,而上述基因变异是胚胎发育不良性神经上皮肿瘤、形成菊形团的胶质神经元肿瘤(RGNT)及其他低级别神经上皮肿瘤的分子遗传学特征。故2020年c IMPACT-NOW更新6将其拟定为独立的肿瘤类型,并在新版肿瘤分类中作为胶质神经元和神经元肿瘤的独立类型。全基因组DNA甲基化分析显示,黏液样胶质神经元肿瘤与其他中枢神经系统肿瘤的甲基化特征有所差异,其特征性基因变异为PDGFRA基因密码子385位点的二核苷酸突变,导致编码血小板源性生长因子受体-α(PDGFR-α)的赖氨酸替换为亮氨酸或异亮氨酸(p.K385L/I)。因此,有研究者将黏液样胶质神经元肿瘤命名为黏液样胶质神经元肿瘤,PDGFRAp.K385突变型[5]。
黏液样胶质神经元肿瘤的组织学形态与胚胎发育不良性神经上皮肿瘤或形成菊形团的胶质神经元肿瘤相似,可见少突胶质细胞样细胞增生,胞核形态一致,呈圆形或卵圆形,核仁小,胞质空亮或轻度嗜酸性;黏液样间质明显,局部可见单个神经元漂浮在黏液样间质中,有时可见嗜碱性黏蛋白的微囊形成;间质内可见细小毛细血管网,缺乏肾小球样血管内皮细胞增生。此外,部分可见神经细胞“菊形团”结构,肿瘤细胞围绕在Syn阳性的嗜酸性神经元纤维核心周围;个别还可见毛细胞样特征以及Rosenthal纤维和嗜酸性小球结构。肿瘤细胞形态温和,核分裂象不明显(<1/10个高倍视野),缺乏钙化和出血坏死灶。免疫组化染色,少突胶质细胞样肿瘤细胞表达GFAP、Olig-2和Y染色体性别决定区框蛋白10(SOX-10);背景的神经轴索和神经毡以及漂浮的神经元表达NF和Syn,少突胶质细胞样肿瘤细胞NF和Syn呈阴性或弱阳性,不表达NeuN和CD34,病变中也未见CD34+细胞(图3)。
黏液样胶质神经元肿瘤若发生于透明隔常伴梗阻性脑积水,若位于胼胝体和脑室周围白质则无脑积水症状。肿瘤生长缓慢,虽然有数例患者发生脑室播散或局部复发,且大多数患者仅行组织活检或次全切除而未予辅助治疗,但临床随访均生存。
三、有少突胶质细胞瘤样特征和核簇的弥漫性胶质神经元肿瘤
有少突胶质细胞瘤样特征和核簇的弥漫性胶质神经元肿瘤(DGONC)是通过全基因组DNA甲基化分析于2020年发现的新肿瘤类型。组织学形态与少突胶质细胞瘤相似,伴肿瘤细胞核簇现象和第14号染色体单体的分子遗传学特征,同时缺乏其他胶质神经元肿瘤的常见分子遗传学变异[6]。新版肿瘤分类列为胶质神经元和神经元肿瘤的暂定类型。
目前报道的有少突胶质细胞瘤样特征和核簇的弥漫性胶质神经元肿瘤十分罕见,主要发生于儿童,发病年龄2~75岁、中位年龄9岁,无性别差异,病变主要位于大脑半球,以颞叶最为多见。影像学表现为幕上界限清晰的皮质或皮质下肿物,无明显的瘤周水肿,T2WI和FLAIR成像呈高信号,增强扫描强化不明显。组织学形态呈弥漫性生长方式,由中至高密度的核周带空晕的圆形肿瘤细胞弥漫分布构成,类似于少突胶质细胞瘤或中枢神经细胞瘤,可见特征性胞核簇集,这些胞核紧密排列在一起,胞质较少,类似于“丢在盘中的硬币(pennies on a plate)”[6],还可见巨细胞、神经毡样岛和围血管假“菊形团”结构以及淋巴细胞浸润和泡沫细胞反应(图4),亦可见核分裂象、灶性坏死和血管内皮细胞增生,因此明确诊断前曾有病例考虑为儿童型间变性少突胶质细胞瘤、非典型脑室外神经细胞瘤,甚至胶质母细胞瘤、高级别神经上皮肿瘤,CNS WHO 4级。免疫组化染色,肿瘤细胞不表达GFAP,但表达神经元标志物微管相关蛋白-2(MAP-2)和Syn,部分表达NeuN,弥漫性表达Olig-2,CD34仅标记血管内皮细胞,而在肿瘤细胞中不表达(图5)。Ki-67抗原标记指数表达不一,个别病例可达30%[7]。约97%的患者存在第14号染色体单体,亦可见少数其他类型变异,如1p获得(26%)、17q获得(58%)和19q缺失(35%),但无其他胶质神经元肿瘤常见的分子遗传学变异。尽管部分肿瘤可复发和进展,但大多数患者经手术切除并术后辅助治疗后预后良好,5年生存率达86%[6]。
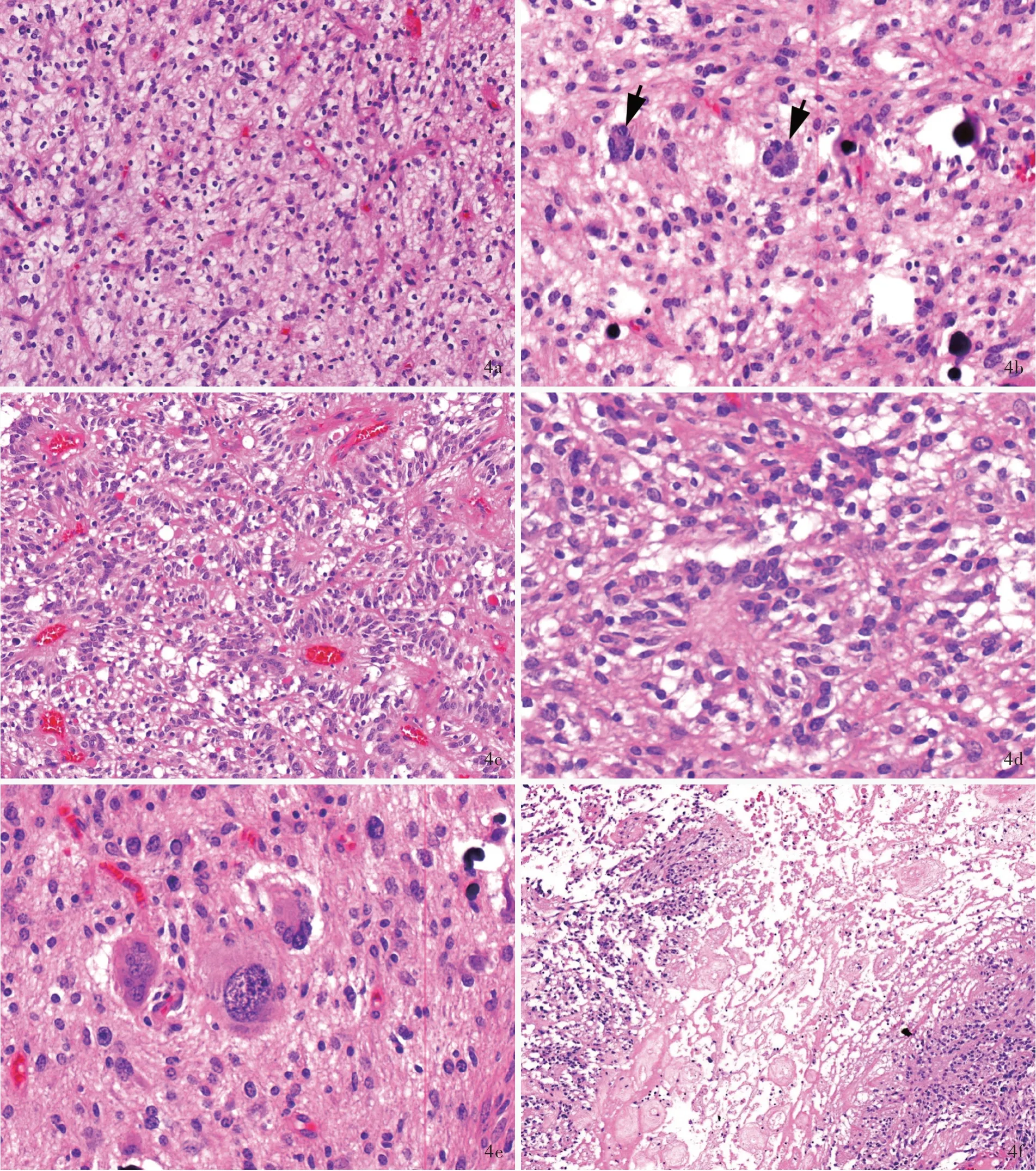
图4 有少突胶质细胞瘤样特征和核簇的弥漫性胶质神经元肿瘤的组织学形态特征 HE染色 4a 肿瘤组织由中至高密度的核周带空晕的圆形肿瘤细胞弥漫分布构成,间质内可见纤细的分支状血管 ×100 4b 可见肿瘤细胞核簇和微钙化灶,胞核紧密排列在一起,胞质较少,类似“丢在盘中的硬币”(箭头所示) ×400 4c 可见部分区域的肿瘤细胞排列呈围血管假“菊形团”结构 ×200 4d 可见神经毡“菊形团”结构 ×400 4e 可见瘤巨细胞 ×400 4f 可见灶性坏死 ×100Figure 4 Histopathological features of DGONC HE staining Tumor showed infiltrating growth pattern with moderate-to highcellularity oligodendroglioma-like tumor cells and delicate branching vessels in stroma(Panel 4a). ×100 Tumor cells had round nuclei with perinuclear haloes and nuclear clusters,resembling"pennies on a plate"(arrows indicate,Panel 4b). ×400 Perivascular pseudorosettes could be observed(Panel 4c). ×200 Neuropil rosettes(Panel 4d)and giant cells(Panel 4e)could be observed. ×400 Tumor necrosis could be encountered in single cases(Panel 4f). ×100
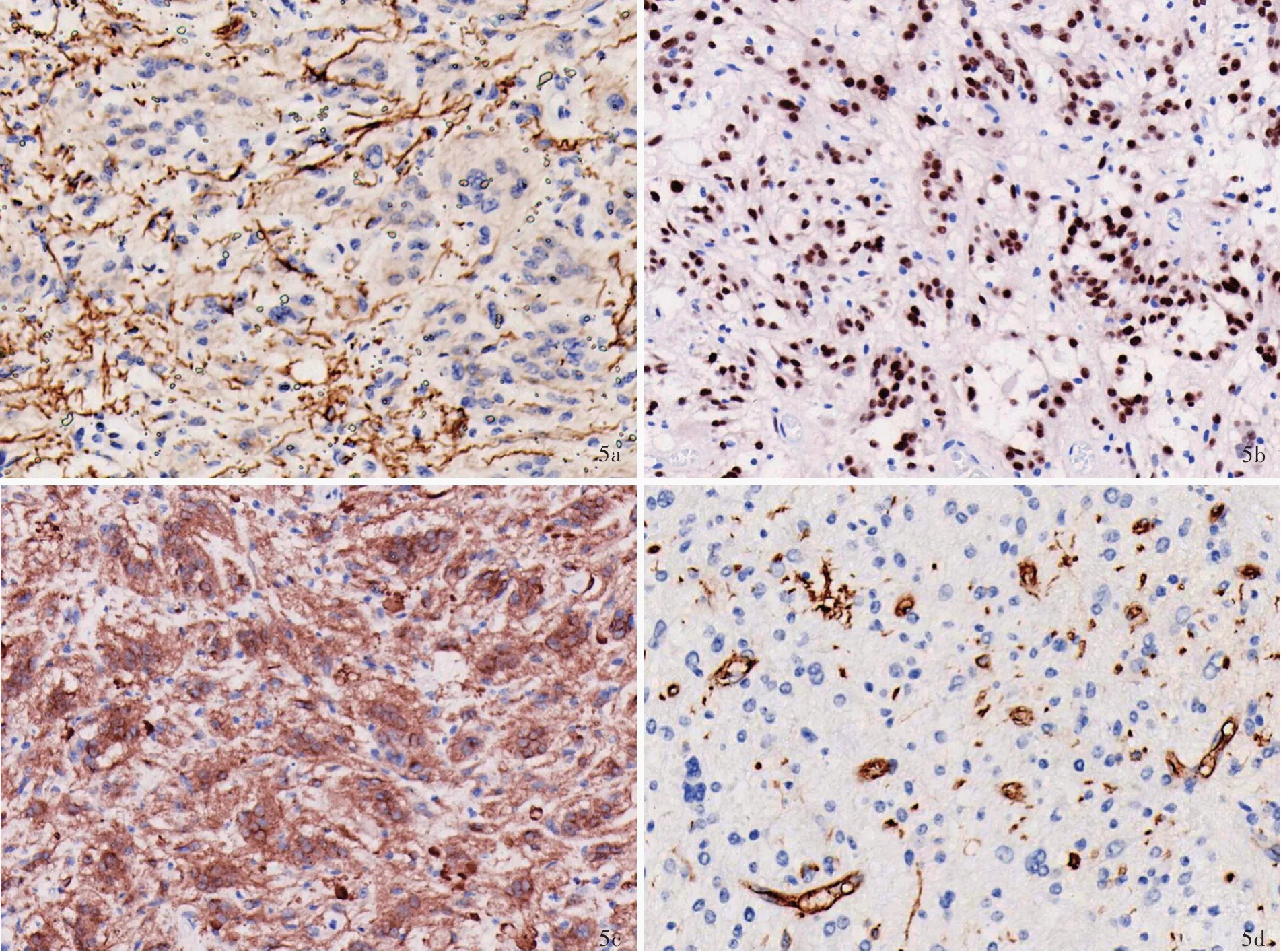
图5 有少突胶质细胞瘤样特征和核簇的弥漫性胶质神经元肿瘤的免疫组化特征 免疫组化染色(EnVision二步法) ×400 5a 肿瘤细胞不表达GFAP,仅可见残留的星形胶质细胞GFAP呈阳性 5b 肿瘤细胞弥漫性表达Olig-2 5c 肿瘤细胞弥漫性表达Syn 5d 仅间质血管内皮细胞表达CD34Figur e 5 Immunohistochemical features of DGONC Immunohistochemical staining(EnVision) ×400 Oligodendroglioma-like tumor cells showed immune-negative for GFAP,only residual astrocytes showed positive for GFAP(Panel 5a),but diffusely positive for Olig-2(Panel 5b)and Syn(Panel 5c).Endothelial cells of blood vessels showed positive for CD34,not in tumor cells(Panel 5d).
四、筛状神经上皮肿瘤
筛状神经上皮肿瘤(CRINET)是发生SMARCB1(INI-1)基因表达缺失的罕见非横纹肌样肿瘤,与非典型性畸胎样/横纹肌样肿瘤(AT/RT)关系密切,于2009年由Hasselblatt等[8]首次报告并命名。组织学形态特殊,可能有相对良好的预后,但能否作为独立的肿瘤类型还是仅作为非典型性畸胎样/横纹肌样肿瘤的特殊亚型尚不确定,故新版肿瘤分类将其列为其他中枢神经系统胚胎性肿瘤的暂定类型。
筛状神经上皮肿瘤好发于婴幼儿,中位发病年龄20个月,无明显性别差异,主要发生于幕上第三脑室近中线处或第四脑室附近以及幕下后颅窝处,术前影像学多考虑脉络丛肿瘤。组织学形态,肿瘤细胞密度较高,由未分化的小细胞形成大小不等的筛孔或厚度不一的梁索状结构,较致密的区域内可见单层肿瘤细胞排列成“菊形团”结构,胞核染色质致密,核仁不明显,胞质轻度嗜酸性,细胞界限不清,可见核分裂象(>2/高倍视野),但缺乏大核仁和核偏位的横纹肌样细胞。免疫组化染色,肿瘤细胞表面表达上皮膜抗原(EMA),局灶性表达细胞角蛋白(CK)和S-100蛋白(S-100),较弥漫性表达波形蛋白(Vim)、MAP-2C、Syn和酪氨酸酶(TYR),不表达或极个别肿瘤细胞表达GFAP,不表达Kir7.1、NF、NeuN、神经元特异性烯醇化酶(NSE)、嗜铬素A(CgA)、平滑肌肌动蛋白(SMA)、结蛋白(Des)、胎盘碱性磷酸酶(PLAP)和人绒毛膜促性腺激素(hCG),Ki-67抗原标记指数可达30%,最重要的是整合酶相互作用分子1(INI-1)表达完全缺失(图6)。筛状神经上皮肿瘤主要表现为染色体22q杂合性缺失和SMARCB1外显子4截短突变以及其他少见的SMARCB1基因变异,亦可见胚系变异[9],这种分子遗传学特征与AT/RT-TYR型相类似,故尚不能确定该肿瘤是否仅为非典型性畸胎样/横纹肌样肿瘤具有组织学特征的亚型。筛状神经上皮肿瘤患者总生存期为100~151个月,中位值为125个月,优于AT/RT-TYR型患者的53个月,可能具有相对良好的预后,但长期预后尚不清楚。
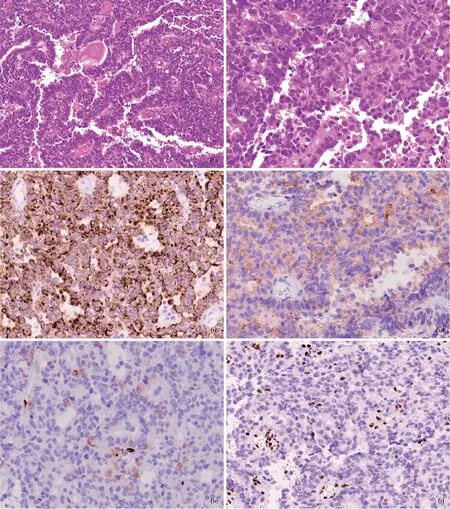
图6 筛状神经上皮肿瘤的病理学特征 6a 肿瘤组织由未分化的小细胞形成厚薄不一的梁索状结构 HE染色 ×200 6b局部形成小腺腔,肿瘤细胞胞核染色质致密,胞质轻度嗜酸性,可见核分裂象,但缺乏大核仁的横纹肌样细胞 HE染色 ×400 6c 肿瘤细胞表达CK(AE1/AE3) 免疫组化染色(EnVision二步法) ×400 6d 肿瘤细胞弱表达Syn 免疫组化染色(EnVision二步法) ×400 6e 极少数肿瘤细胞表达GFAP 免疫组化染色(EnVision二步法) ×400 6f 肿瘤细胞INI-1表达缺失 免疫组化染色(EnVision二步法) ×400Figure 6 Histopathological and immunohistochemical features of CRINET Tumor was characterized by the presence of strands and ribbons with varying thickness(Panel 6a). HE staining ×200 In some areas,small lumina were also present with light eosinophilic cytoplasms and active mitotic figures,but rhabdoid tumor cells with prominent nucleoli were absent(Panel 6b). HE staining ×400 Tumor cells showed positive for CK(AE1/AE3)diffusely(Panel 6c)and Syn weakly(Panel 6d),only scattered cell positive for GFAP(Panel 6e),but negative for INI-1(Panel 6f). Immunohistochemical staining(EnVision) ×400
五、中枢神经系统神经母细胞瘤,FOXR2活化型
中枢神经系统神经母细胞瘤,FOXR2活化型(CNS neuroblastoma,FOXR2-activated)是一种具有神经母细胞和(或)神经细胞分化并具有数目不等神经节细胞和神经毡基质的胚胎性肿瘤,常有染色体1q获得以及通过各种结构重排激活转录因子FOXR2。该肿瘤是2016年Sturm等[10]对中枢神经系统原始神经外胚层肿瘤(CNS PNET)进行全基因组DNA甲基化分析时发现的4个新肿瘤类型之一。新版肿瘤分类将其列为其他中枢神经系统胚胎性肿瘤中的独立类型。
中枢神经系统神经母细胞瘤,FOXR2活化型好发于儿童,影像学可见界限清晰的大脑肿物。发病机制涉及FOXR2复杂的染色体间和染色体内重排,包括JMJD1C-FOXR2融合和多种串联复制,后者将相邻基因的启动子区域并列以激活FOXR2的表达,导致肿瘤FOXR2水平高于其他中枢神经系统肿瘤类型。组织学形态呈现胚胎性结构和小细胞表型的特征,肿瘤细胞小而圆,胞质少,胞核深染,密集增生的肿瘤细胞内可见神经毡、神经细胞或神经节细胞等分化区域,及血管周围无核区(围血管假“菊形团”结构)、胞核“栅栏”状排列和Homer-Wright“菊形团”结构,亦可见间质黏液变性和微血管增生。免疫组化染色,肿瘤细胞FOXR2、NKX2-1、Olig-2和Syn呈弥漫性强阳性,绝大多数细胞不表达GFAP和Vim。由于FOXR2在正常脑组织或其他中枢神经系统肿瘤类型中不表达,故FOXR2强阳性有助于诊断。中枢神经系统神经母细胞瘤的其他基因变异,如有MYC扩增的中枢神经系统神经母细胞瘤,不能诊断为独特类型,仅能诊断为中枢神经系统神经母细胞瘤,NEC。中枢神经系统神经母细胞瘤,FOXR2活化型预后中等,部分病例可生存10年且无复发,为该肿瘤降低治疗强度提供了证据[11]。
六、有BCOR内部串联重复的中枢神经系统肿瘤
有BCOR内部串联重复的中枢神经系统肿瘤(CNS BCOR ITD)是一种高度侵袭性胚胎性肿瘤,也是通过全基因组DNA甲基化分析发现的中枢神经系统原始神经外胚层肿瘤的4个新肿瘤类型之一[10],最初将其命名为有BCOR变异的中枢神经系统高级别神经上皮肿瘤(CNS high-grade neuroepithelial tumor withBCORalteration),2020年cIMPACT-NOW更新6将其拟定为独立的肿瘤类型并更名为有BCOR内部串联重复的中枢神经系统肿瘤。尽管目前尚不清楚其与中枢神经系统外BCOR变异性肿瘤(例如肾透明细胞肉瘤和婴儿原始黏液样间叶性肿瘤等)的关系,考虑是不同祖细胞发生相似基因变异产生的不同肿瘤类型:有BCOR内部串联重复的中枢神经系统肿瘤起源于原始神经上皮祖细胞,中枢神经系统外BCOR变异性肿瘤起源于间叶性祖细胞。因此,建议将有BCOR内部串联重复的中枢神经系统肿瘤归类为其他胶质瘤,新版肿瘤分类将其列为其他中枢神经系统胚胎性肿瘤的独立类型。
有BCOR内部串联重复的中枢神经系统肿瘤好发于儿童,可发生于中枢神经系统任意部位,小脑肿瘤仅见于<5岁幼儿,幕上肿瘤则好发于幼儿和年龄较大儿童。组织学形态复杂多样,可表现为形态单一的圆形或卵圆形细胞实体性片状分布,内含丰富的纤细薄壁血管网;可由胞质嗜酸性、具有胶质纤维的多形性神经胶质细胞或少突胶质细胞样有核周空晕的细胞构成,并形成间质内“鸡笼”样血管网;还可表现为胞核深染的胚胎性小细胞密集片状分布。部分以室管膜瘤样围血管“菊形团”结构和乳头状结构为主。个别可见组织学异质性,部分区域为组织学低级别,部分区域为显著的间变特征,可能与间变区域同时伴TP53基因突变有关。可见假“栅栏”状坏死,但缺乏微血管增生。免疫组化染色,肿瘤细胞全部或绝大部分不表达GFAP,仅少量反应性星形胶质细胞GFAP呈阳性;Olig-2表达不一,可完全阴性或小灶性阳性;大多数肿瘤细胞表达NeuN(10%~80%);不表达Syn;胞质散在表达NF;EMA呈阴性或胞质呈颗粒状弱阳性,缺乏室管膜瘤样核旁点状阳性;胞核BCOR呈弥漫性强阳性;Ki-67抗原标记指数15%~60%[12]。分子病理学方面,具有独特的BCOR外显子15内部串联重复杂合性变异(野生型等位基因并未丢失),但部分病例除上述特征性分子遗传学改变外,还伴CDKN2A/B纯合性缺失、TERT扩增或启动子突变以及TP53、BCORL1、EP300、SMARCA2、STAG2变异。尽管多数病例提示有BCOR内部串联重复的中枢神经系统肿瘤预后较差,但也有个别病例长期生存,其机制尚待进一步探讨。
七、松果体区促纤维增生性黏液样肿瘤,SMARCB1突变型
松果体区促纤维增生性黏液样肿瘤,SMARCB1突变型(desmoplastic myxoid tumor of the pineal region,SMARCB1-mutant)是2020年Thomas等[13]新发现并命名的位于松果体区具有独特组织学形态特点的肿瘤性病变,由分布于疏松黏液样基质中的上皮样、梭形肿瘤细胞和明显胶原化的促纤维增生间质交替分布构成,同时具有SMARCB1基因纯合性或杂合性缺失的分子遗传学特征。新版肿瘤分类将其列为松果体肿瘤的新增独立类型。
截止目前,采用松果体区促纤维增生性黏液样肿瘤,SMARCB1突变型命名的肿瘤不超过10例报道[13-15],均发生于松果体区,但实际上肿瘤可能并不局限于此,有2例组织学形态特征相似的病变发生于脑膜,认为是脑膜原发性SMARCB1缺失性肿瘤(primary meningealSMARCB1-deficient tumors)[16],故尚待更多的病例资料支持或取消该肿瘤的部位特殊性。
松果体区促纤维增生性黏液样肿瘤,SMARCB1突变型多发生于成人,平均发病年龄40岁,无性别偏向性。组织学形态可见卵圆形、梭形或上皮样肿瘤细胞分布于促纤维增生的间质背景中,胶原化纤维条索与疏松黏液样基质交替混杂分布,但更普遍的是,促纤维增生区域与黏液样区域分区存在。大部分病例可见玻璃样变性的硬化胶原束,但胞核大、胞质丰富的横纹肌样细胞仅见于个别病例。肿瘤细胞形态较温和,胞质嗜酸性或淡染,核分裂象少见(<1/20个高倍视野),缺乏肿瘤坏死,部分病例可见淋巴细胞在间质内浸润。免疫组化染色,肿瘤细胞INI-1完全缺失,大部分病例表达CD34、EMA和Vim,局灶性表达Syn和SMA,不表达CK和GFAP;部分病例表达Brachyruy,应注意与脊索瘤相鉴别;Ki-67抗原标记指数偏低(约3%),个别病例可达15%(图7)。全基因组DNA甲基化分析结果与AT/RT-MYC型和低分化脊索瘤相近,表明该肿瘤与非典型性畸胎样/横纹肌样肿瘤关系密切,但预后优于儿童或成人非典型性畸胎样/横纹肌样肿瘤。由于目前积累的病例较少,二者之间的关系尚待进一步阐明。
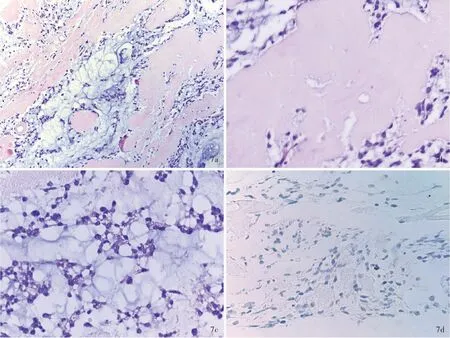
图7 松果体区促纤维增生性黏液样肿瘤,SMARCB1突变型的病理学特征 7a 肿瘤组织由富含黏液的区域和玻璃样变性的促纤维增生区域交替分布构成 HE染色 ×100 7b 在促纤维增生的胶原化区域内细胞成分较少,胶原组织明显玻璃样变性HE染色 ×400 7c 在富含黏液的区域内可见圆形或卵圆形胞核,胞质嗜酸性上皮样或梭形细胞散在分布,肿瘤细胞形态温和 HE染色 ×400 7d 肿瘤细胞INI-1完全缺失 免疫组化染色(EnVision二步法) ×400Figur e 7 Histopathological and immunohistochemical features of desmoplastic myxoid tumor of the pineal region,SMARCB1-mutant Tumor exhibited admixture of variably dense desmoplastic area of hyaline degeneration and tumor cells embedded in a loose pale basophilic myxoid matrix(Panel 7a). HE staining ×100 Desmoplastic area of tumor showed hypocellular with a prominent hyalinized,fibrosclerotic matrix(Panel 7b). HE staining ×400 Small-to middle-sized oval to spindled and epithelioid cells with bland appearance embedded in loose pale basophilic myxoid matrix(Panel 7c). HE staining ×400 Tumor cells showed loss of nuclear INI-1 protein expression(Panel 7d). Immunohistochemical staining(EnVision) ×400
八、CIC重排肉瘤
CIC重排肉瘤(CIC-rearranged sarcoma)是于2020年WHO公布的骨和软组织肿瘤分类(第五版)中新增的肿瘤类型,组织学形态类似尤文肉瘤的小圆蓝细胞肿瘤,但缺少EWSR1易位,呈现染色体19q13的CIC重排。主要发生于青年,多位于骨外的四肢和躯干深部软组织,罕见发生于骨骼、内脏和颅内。颅内CIC重排肉瘤的诊断应遵循软组织肿瘤的原则,发生于骨和软组织者最常见的融合伙伴基因 为DUX4(95%),其 次 为FOXO4、NUTM2A和NUTM1等,大多数发生于颅内者的伙伴基因为NUTM1[10]。目前尚不清楚CIC不同融合伙伴基因是否影响肿瘤的生物学行为和临床预后。
CIC重排肉瘤的组织学形态与尤文肉瘤相似,通常由小至中等大小的圆形肿瘤细胞构成,胞核深染或空泡状,核仁明显,胞质嗜双色至淡嗜酸性,肿瘤细胞通常排列成分叶状,可见较广泛的“地图”样坏死,核分裂象和病理性核分裂象易见,胞核多形性较经典的尤文肉瘤更明显。少数病例可见透亮细胞、梭形细胞和黏液样基质,偶见上皮样细胞或横纹肌样细胞特征。CIC-NUTM1重排肉瘤组织学形态表现为弥漫性黏液样基质,可见圆形细胞区域和梭形细胞区域,类似肌上皮瘤样生长模式。免疫组化染色,大多数胞核弥漫性表达DUX4,Fli-1和CD99阳性,而CD99呈局灶性阳性而非尤文肉瘤的胞膜弥漫性阳性。因此,若小圆形细胞肿瘤局灶性表达或不表达CD99时,应考虑CIC重排肉瘤。大部分病例表达ETV4、WT1和钙网蛋白(calreticulin),少数病例表达CK(AE1/AE3)、Des、S-100和ERG,不表达NKX2.2。应注意的是,CIC-NUTM1重排肉瘤弥漫 性表达NUT,亦 表达CK(AE1/AE3)、SOX-2、WT1和ETV4,而不表达S-100、P63和P40,须与NUT癌相鉴别。此外,采用CIC分离探针行荧光原位杂交(FISH)时,假阴性率(隐性重排)约15%[17],需第二代测序技术(NGS)进一步验证。
CIC重排肉瘤常表现为播散性疾病进展,有较强的化疗抵抗性,总生存率低于尤文肉瘤(5年生存率仅43%,尤文肉瘤为77%)。CIC-NUTM1重排肉瘤多发生于颅内,总生存期为7~37个月,中位值17.5个月。由此可见,CIC-NUTM1重排肉瘤的预后更差,尚待更多病例总结临床病理学特征和肿瘤生物学行为。
九、颅内间叶性肿瘤,FET-CREB融合阳性型
颅内间叶性肿瘤,FET-CREB融合阳性型(intracranial mesenchymal tumor,FET-CREB fusion-positive)是新近定义的具有特殊分子遗传学特征的肿瘤类型。FET(FUS/EWSR1/TAF15)家族成员均为RNA结合蛋白,在多种软组织肉瘤中可发生染色体易位致新融合蛋白形成。转录因子CREB家族包括重要的核转录因子ATF1、CREB1和CREM。软组织血管瘤样纤维组织细胞瘤、软组织透明细胞肉瘤、胃肠道透明细胞肉瘤、原发性肺黏液样肉瘤、涎腺玻璃样透明细胞癌等均存在这两个家族成员间的基因融合。颅内FET-CREB融合肿瘤分别描述为颅内血管瘤型纤维组织细胞瘤(AFH)、颅内血管瘤型纤维组织细胞瘤,黏液亚型或颅内黏液样间叶性肿瘤(IMMT),主 要涉及EWSR1、FUS与ATF1、CREB1、CREM间的融合[18-19]。尽管具有较特殊的组织学形态和分子遗传学特征,但目前仍不确定颅内血管瘤型纤维组织细胞瘤和颅内黏液样间叶性肿瘤是一种疾病还是两个独立肿瘤类型,也不明确颅内间叶性肿瘤,FET-CREB融合阳性型是特定肿瘤的黏液样亚型还是独立的肿瘤类型[20],故新版肿瘤分类将其列为未确定分化的肿瘤的暂定类型。
颅内间叶性肿瘤,FET-CREB融合阳性型主要发生于青年人,高峰发病年龄为20~30岁,但年龄跨度较大,4~70岁均可发生;好发于脑实质外硬脑膜,少数发生于脑室内;影像学表现为界限清晰的结节,呈分叶状,有实性和囊性成分,增强扫明呈明显强化,部分可见“脑膜尾征”和颅骨侵犯,个别沿软脑膜播散。组织学形态表现出较宽的形态学谱系,且不同基因融合各有其独特表现。所有融合类型肿瘤均可见胶原化间质背景,有时纤维间隔可将肿瘤细胞分隔成结节状,有时背景是丰富的黏液样基质。肿瘤细胞呈上皮样、横纹肌样、星芒状、梭形和圆形,其中星芒状和梭形细胞最常见于丰富的黏液样基质中,亦可见上皮样细胞,类似脊索样脑膜瘤。另一较普遍的表现为,大多数病例可见肿瘤周围或纤维间隔处密集的淋巴浆细胞集聚,以及瘤内出血伴含铁血黄素沉积,但缺乏颅外软组织血管瘤型纤维组织细胞瘤的特征性假性血管瘤样腔隙。上皮样或横纹肌样细胞形态在EWSR1-ATF1融合的肿瘤中更常见,而EWSR1-CREB1融合的肿瘤更多见富黏液的间质、星芒状和梭形细胞。大部分核分裂象较低(<5个/mm2),但FUS-CREM融合的肿瘤通常由密集排列的上皮样和横纹肌样细胞构成,具有显著的细胞异型性和活跃的核分裂活性[21]。免疫组化染色,肿瘤细胞表达Des,呈弥漫性强阳性或局灶性弱阳性;大多数病例EMA和CD99呈膜状阳性,部分表达S-100、Syn、MUC4和CD68,个别表达CK(AE/AE3)、CAM5.2和Pan-NTRK,但 是 不 表 达GFAP、Myogenin、生 长 抑 素 受 体2(SSTR2)和HMB45,INI-1表达不缺失,Ki-67抗原标记指数为15%~20%。特殊染色可见细胞间存在丰富的网状纤维。其分子病理学的主要变异包括EWSR1-ATF1融合(最多见)、EWSR1-CREB1融合、EWSR1-CREM融合和FUS-CREM融合(最少见),也伴少量染色体获得或缺失,但无明显的基因扩增、突变和缺失等变异。颅内间叶性肿瘤,FET-CREB融合阳性型有局部复发倾向,偶尔播散或转移致死亡,是否不同基因融合导致预后不同,尚待进一步研究。
十、原发性颅内肉瘤,DICER1突变型
原发性颅内肉瘤,DICER1突变型(primary intracranial sarcoma,DICER1-mutant)是一种原发于颅内的间叶源性肉瘤,由梭形或卵圆形细胞、有时是多形性细胞构成,伴横纹肌母细胞或横纹肌母细胞样细胞的特殊形态以及DICER1基因变异的分子遗传学特征。最初由Koelsche等[22]于2018年描述并将其命名为有横纹肌肉瘤样特征的梭形细胞肉瘤,DICER1突变型(spindle cell sarcoma with rhabdomyosarcoma-likefeatures,DICER1mutant)。随后报道的病例中出现以多形性肿瘤细胞为主的组织学形态特点,故提议将其命名为原发性颅内肉瘤,DICER1突变型[23]。新版肿瘤分类接受这一命名,并将其列为未确定分化的肿瘤的新增类型。
DICER1基因定位于第14号染色体q32.13,编码核糖核酸酶Ⅲ(RNaseⅢ)家族的核糖核酸内切酶DICER,后者直接参与RNA干扰(RNAi)途径,调控基因表达的后续翻译。DICER1基因变异可导致微小RNA(miRNA)功能失调,使抑癌基因功能缺失或致癌基因功能增强,从而诱发多种肿瘤的发生。原发性颅内肉瘤,DICER1突变型好发于儿童,平均发病年龄6岁,个别可见于成人和老年人,无性别偏向性,主要位于幕上。组织学形态,大多数肿瘤由密集增生的梭形和卵圆形细胞弥漫分布构成,部分区域肿瘤细胞呈束状或“车辐”状排列,少数由多形性肿瘤细胞弥漫分布构成,胞核多形性和多核瘤巨细胞,可见明显大小不等的胞质内嗜酸性小球,亦可见胞质红染、胞核偏位的横纹肌母细胞或横纹肌母细胞样细胞,这些细胞多位于肿瘤细胞较疏松的区域,呈簇状分布,密集区域也可见散在的横纹肌母细胞;核分裂象活跃,可见病理性核分裂象;可见肿瘤细胞局灶性凝固性坏死,少数还可见黏液样区域和软骨分化。免疫组化染色,肿瘤细胞弥漫性表达CD99和Vim,部分表达肌源性标志物Des、肌动蛋白(HHF-35)、Myogenin和Myoglobin,高表达断裂转导蛋白样增强子1(TLE1)和P53,部分病例ATRX表达缺失,SMA表达不一,H3 K27me3呈“马赛克”样表达、部分表达缺失。有研究显示,TLE1和H3 K27me3的表达特征可以高度提示原发性颅内肉瘤,DICER1突变型的诊断[24]。肿瘤细胞不表达CK、GFAP、Olig-2、SOX-2、SOX-10、Syn、CAM5.2、EMA、CD34、信号转导与转录激活因子6(STAT6)、SSTR2A、间变性淋巴瘤激酶(ALK),无INI-1和BRG1表达缺失,Ki-67抗原标记指数较高(>50%)。特殊染色显示肿瘤细胞间有丰富的网状纤维。分子病理学检测,存在DICER1基因1种或多种致病性变异,可为胚系变异或体细胞变异,包括DICER1RNaseⅢb热点突变和缺失,还可伴其他基因如TP53、ATRX、MAPK通路等胚系变异或体细胞变异。由于目前病例数较少,对其临床预后尚不清楚。
十一、垂体母细胞瘤
垂体母细胞瘤(PitB)是极为罕见的腺垂体肿瘤,由Scheithauer等[25]于2008年首先报告并命名,于2017年被WHO内分泌肿瘤分类列为新增肿瘤类型[26],在新版肿瘤分类中新增为鞍区肿瘤类型。垂体母细胞瘤属于神经内分泌肿瘤范畴,目前全世界仅报道30余例,几乎所有病例均发生于24个月内的婴幼儿,临床可表现为库欣综合征的满月脸、水牛背,也可为神经性眼病和尿崩症等,血浆或血清促肾上腺皮质激素(ACTH)水平升高,其他垂体激素水平亦受不同程度的影响。影像学表现为鞍区或鞍上囊实性或实性肿物。
组织学形态,垂体母细胞瘤重现垂体各细胞发育阶段,组织结构与胚胎期垂体类似,包括3种细胞成分,即原始胚芽细胞呈小叶状分布,细胞小,胞核深染;类似未成熟Rathke柱状上皮的上皮细胞成分,细胞呈矮立方柱状,可形成“菊形团”或腺腔样结构;体积稍大的分泌细胞,呈圆形或者卵圆形,胞质丰富淡染,核仁不明显,类似腺垂体腺泡细胞,其间夹杂滤泡星状细胞。核分裂活性各异,可见坏死区域。免疫组化染色,分泌细胞表达Syn、CgA等神经内分泌标志物,大多数病例不同程度表达ATCH和生长激素(GH),不表达或者仅局灶性弱表达其他腺垂体激素;散在的滤泡星状细胞少量表达GFAP和S-100,腺上皮细胞表达CK、CK8/18、Galectin-3;原始胚芽细胞表达P27和P53;Ki-67抗原标记指数不一。全基因组DNA甲基化分析显示,垂体母细胞瘤有独特的甲基化谱系,不同于其他垂体肿瘤,如垂体腺瘤、神经垂体肿瘤等,存在DICER1基因1种或多种致病性变异(胚系变异或体细胞变异以及RNaseⅢ热点突变等)[27],提示垂体母细胞瘤可能是DICER1相关综合征的成员。由于肿瘤生长部位的特殊性,垂体母细胞瘤病死率较高,但手术全切除和长期内分泌治疗可使患者获得较好预后和较高的生活质量[28]。
新版肿瘤分类新增肿瘤类型反映了对肿瘤临床病理学和分子遗传学特征的最新认知,其目的是加强对肿瘤生物学行为的理解和提高诊断的准确性,并最终完善治疗方案,使患者获益。尽管在新增肿瘤类型的确定过程中,特征性分子遗传学变异的发现发挥了重要作用,但组织学特征仍然是精确分类的关键。
志谢 感谢中山大学孙逸仙纪念医院病理科卞丽娟和王琳医生、中山大学肿瘤防治中心病理科胡婉明医生、北京市神经外科研究所神经病理室刘幸医生提供部分病例图片
利益冲突 无
猜你喜欢
肝博士(2021年1期)2021-03-29 02:32:08
家庭医学(下半月)(2019年11期)2020-01-16 08:39:08
家庭医学(下半月)(2019年10期)2019-11-16 08:59:52
中国临床医学影像杂志(2019年5期)2019-08-27 02:48:00
中国临床医学影像杂志(2019年4期)2019-06-18 10:55:06
中国临床医学影像杂志(2019年2期)2019-04-25 06:15:50
现代检验医学杂志(2016年3期)2016-11-15 01:59:26
中国中西医结合皮肤性病学杂志(2016年4期)2016-07-18 10:59:56
成都中医药大学学报(教育科学版)(2016年1期)2016-01-22 07:24:50
实用肝脏病杂志(2015年5期)2015-12-03 06:28:04
