
非晶态TiB2@C催化剂对NaAlH4合成和可逆储氢性能的影响
2021-02-01 01:36曹志杰李丽江马晓波
西安交通大学学报 2021年1期
曹志杰,李丽江,马晓波
(宁夏大学物理与电子电气工程学院,750021,银川)
固态储氢材料具有储氢密度高、稳定性好、安全性高、运输方便等优点,是近年来最具发展潜力的车载储氢材料之一[1-3]。为了寻找合适的车载储氢材料,研究人员做了大量工作,开发出包括低容量过渡金属氢化物(AB、AB2、AB5)和高容量配位氢化物(铝氢化物、硼氢化物、氨基氢化物)等多种储氢材料[4]。轻金属配位氢化物NaAlH4由于具有理论储氢量高、前两步放氢反应的热力学条件温和、从原料NaH和Al制备成本低等优势而受到广泛关注[5],但是可逆性受限、放氢温度偏高、吸放氢动力学缓慢等问题仍然制约着NaAlH4的实际应用。
围绕NaAlH4的上述问题,研究人员开发出多种改性方法,包括掺杂催化剂[6]、纳米化[7]、反应去稳定[8]等。Bogdanovic等通过添加β-TiCl3和Ti(OBun)4,首次实现了NaAlH4的可逆吸放氢,这一研究工作使得掺杂Ti基催化剂改性备受关注[9]。Sandrock等研究发现,通过球磨掺杂TiCl3的NaAlH4体系在125 ℃下100 min内的放氢量(质量分数)为3%,随后在相同温度、9.1 MPa氢压下1 h内的吸氢量高达4.2%[10]。Wang等比较了TiCl3和TiF3对NaAlH4放氢性能的影响,发现TiF3的催化效果优于TiCl3:掺杂TiF3的样品放氢后在60 ℃、11 MPa氢压下6 h内的吸氢量为3.4%,而掺杂TiCl3的样品放氢后在80 ℃、11 MPa氢压下10 h内的吸氢量仅为3.2%[11]。对于这些催化体系而言,最大的问题在于吸放氢过程中Ti基催化剂会与NaAlH4发生反应并生成惰性副产物,从而降低复合体系的实际储氢量[12]。因此,许多研究开始转向在反应过程中能够保持稳定的Ti基催化剂。Li等在NaAlH4中掺杂纳米TiN后,复合体系的起始放氢温度由160 ℃降低至130 ℃,第一步放氢的峰值温度由198.9 ℃降低至183.5 ℃[13]。Li等研究了TiB2掺杂对NaAlH4的催化效果,热力学分析表明掺杂体系的起始放氢温度低至75 ℃,在190 ℃下1 h内的放氢量为3.60%,4 h内的放氢量则达到了5.21%,显示出优异的放氢动力学性能[14]。邱昊辰等在CeH2掺杂NaAlH4的体系中添加Ti31Cr15.5V45Fe8.5Ce0.5,当合金的原子分数达到30%时,样品在150 ℃下的放氢量从1.89%上升至2.31%,第一步放氢反应时间降低了17 min,放氢速率提高了33.81%,结果表明两种催化剂共掺杂的效果要比单一的催化剂优异[15]。为了增强催化效果,Zhang等制备出TiO2颗粒分散于碳基体上的TiO2和C的复合催化剂(简写为TiO2@C),研究表明掺杂9%的TiO2@C后,NaAlH4复合体系在140 ℃下10 min内的放氢量高达4.2%,并且放氢后的样品在10 MPa氢压下能够吸氢实现完全可逆[16]。Cheng等研究表明,采用NbF5和有序纳米碳管(CMK-3)共掺杂体系比单独添加NbF5或CMK-3体系的放氢温度低,共掺杂体系前两步放氢过程的活化能显著降低,并表现出优异的循环稳定性[17]。
上述研究表明,采用不同催化剂共掺杂能够有效地改善NaAlH4的综合储氢性能。为了结合TiB2和碳的催化效果,本工作制备出TiB2分散于碳基体上的非晶态TiB2@C复合催化剂,并以NaH和Al为原料,在氢气气氛下采用球磨法一步合成NaAlH4,着重研究TiB2@C掺杂对NaAlH4的合成以及复合体系可逆吸放氢性能的影响。
1 实验过程
1.1 TiB2@C的制备
TiB2@C的制备过程如图1所示:采用市售的二茂钛二氯化物(纯度97%)和硼氢化锂(纯度大于等于98%)为原材料,均匀混合后装入不锈钢球磨罐中,然后置于行星式球磨机(MM500,Retsch)上球磨3 h,转速为400 r/min,球料质量比为40∶1。然后,在手套箱中将球磨产物密封于石英管,并在550 ℃、氩气气氛保护下烧结2 h,加热速率为5 ℃/min,H2O和O2的体积分数均低于10-6。烧结过程中发生如下反应:Cp2TiCl2+2LiBH4→TiB2+C+2LiCl[18]。采用乙醇将烧结产物清洗三遍以除去副产物(LiCl),清洗后的样品在80 ℃下真空干燥8 h,得到目标催化剂。

图1 TiB2@C制备过程示意图
1.2 TiB2@C掺杂NaAlH4复合体系的制备
将氢化钠(纯度97%)和铝粉(纯度99.5%)混合均匀装入不锈钢球磨罐,并加入6%的TiB2@C,球磨罐密封后充入5 MPa氢气,并置于行星式球磨机上进行球磨,转速为400 r/min,球料质量比为40∶1,球磨程序设定为每旋转30 min中间停留10 min。为了比较,还在相同条件下球磨了未添加催化剂的NaH/Al混合物以及纯NaAlH4。球磨前后,所有的样品处理过程均在手套箱中进行。
1.3 样品的结构及性能表征
采用X射线衍射技术(XRD)对样品的相结构进行分析,采用Stoe STADI P透射衍射仪。XRD测试时,所有制备的样品均封装于氩气气氛保护的毛细玻璃管中,玻璃管直径为0.5 mm。采用透射电子显微镜(TEM)和扫描电子显微镜(SEM)对催化剂的形貌和成分进行分析,仪器型号为Hitachi S-5500。采用傅里叶红外光谱仪(FT-IR)分析样品的化学键信息,仪器型号为Nicolet IS50。热重分析(TGA)和差示扫描量热法(DSC)的测量是在氩气气氛保护下的TGA/DSC STARe仪器上进行,同时采用质谱仪(MS,HPR-20 QMS)对放氢过程进行检测,加热速率均为5 ℃/min。采用程序升温脱附(TPD)系统分析样品的放氢过程,升温速率为5 ℃/min。
2 结果与讨论
2.1 TiB2@C的结构特征
图2为TiB2@C催化剂的XRD图谱。由图可知,Cp2TiCl2和LiBH4发生反应后,产物中除了副产物LiCl的微弱衍射峰之外并没有出现其他物相的衍射峰,同时在20°~25°之间出现了馒头峰。采用乙醇清洗三次后,样品中LiCl的质量分数减少,其衍射峰消失,而馒头峰保持不变,说明所制备的催化剂中TiB2和C可能处于非晶状态。
图3为TiB2@C催化剂的TEM、SEM和EDS面扫描图片。由TEM可知,样品中并没有出现明显的颗粒状物质,高分辨照片也未见晶格条纹,说明样品中TiB2和C均处于非晶态。由SEM可知,样品呈现无规则絮状。EDS面扫描结果显示,Ti和C都均匀分布在样品中,说明催化剂由非晶态TiB2和无定形碳组成,并且TiB2均匀分布在无定形碳基体中,因此本文中将催化剂标记为TiB2@C。此外,还有少量的Cl元素遗留在样品中,说明乙醇清洗并没有完全除去副产物LiCl。

(a)低倍透射电镜照片(b)高倍透射电镜照片
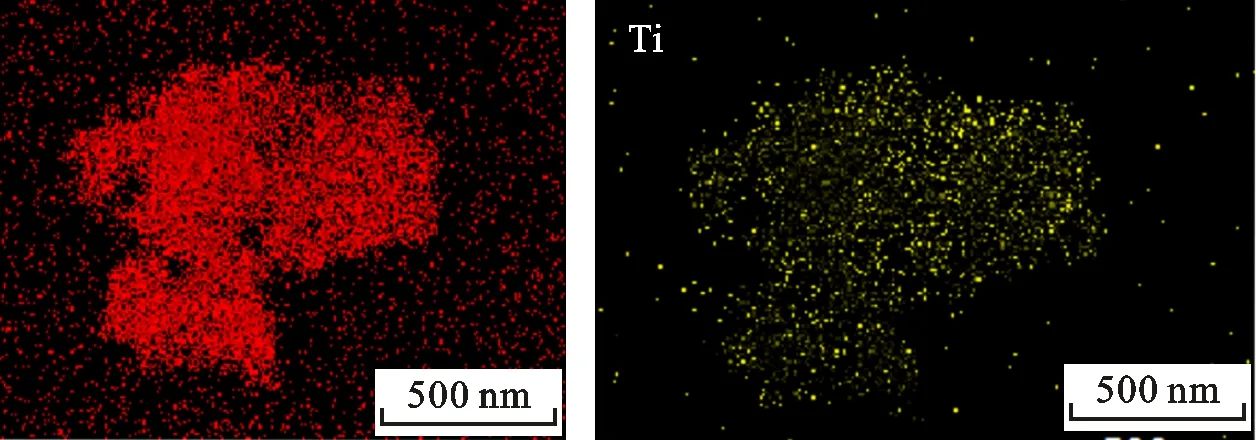
(c)扫描电镜照片(d)Ti的面扫描图片

(e)C的面扫描图片(f)Cl的面扫描图片图3 TiB2@C催化剂的透射电镜照片、扫描电镜照片和Ti、C、Cl的面扫描图片
2.2 TiB2@C添加对NaAlH4合成和吸放氢性能的影响
为了考察TiB2@C对合成NaAlH4的催化效果,在NaH和Al的混合物中添加6%的TiB2@C并置于氢气气氛下球磨,球磨产物的XRD如图4所示。球磨5 h后,产物中出现了Na3AlH6,并随着球磨时间的延长,其衍射峰的强度逐渐增大,同时NaH和Al的强度逐渐减弱,说明在球磨过程中发生了如下反应:3NaH+Al+1.5H2→Na3AlH6。球磨20 h后,NaH相消失,样品中只剩下Na3AlH6和Al的衍射峰。随着球磨时间的进一步延长,Na3AlH6和Al的衍射峰强度逐渐减弱,产物中出现了NaAlH4且其衍射峰强度逐步增强,说明反应按照Na3AlH6+2Al+3H2→3NaAlH4的路径进行。当球磨时间达到50 h时,Na3AlH6和Al相均消失,产物中只剩下NaAlH4的衍射峰,说明在球磨过程中NaH和Al完全转变成了NaAlH4,由于球磨过程中添加了6%的TiB2@C作为催化剂,球磨产物标记为NaAlH4-6%TiB2@C。

图4 NaH/Al-6%TiB2@C在氢气气氛下球磨不同时间的XRD图谱
为了比较,在相同条件下分别球磨了NaAlH4-6%TiB2@C、NaAlH4、NaH和Al混合物,球磨后的XRD图谱如图5所示。球磨50 h后,纯NaAlH4的物相保持不变,而未添加TiB2@C的NaH/Al混合物,只生成了少量Na3AlH6。对比NaAlH4-6%TiB2@C,反应物NaH和Al全部变化生成了NaAlH4,说明在氢气气氛下球磨过程中TiB2@C能够有效地促进NaH和Al原位氢化生成NaAlH4。

图5 NaAlH4,NaH/Al和NaH/Al-6%TiB2@C在相同条件下球磨50 h后的XRD图谱
2.3 TiB2@C添加对NaAlH4吸放氢性能的影响
图6为NaAlH4-6%TiB2@C和NaAlH4的TGA、DSC及MS曲线。由图6中TGA曲线可知,TiB2@C的添加明显降低了NaAlH4的放氢温度:NaAlH4的起始放氢温度约为18 ℃,而NaAlH4-6%TiB2@C的起始放氢温度降低至10 ℃。NaAlH4-6%TiB2@C前两步放氢过程(100~210 ℃)的放氢量约为4.9%,相比于球磨NaAlH4,其放氢量有所降低,但具有更好的综合放氢性能。由图6中DSC曲线可知,NaAlH4在40~400 ℃范围内出现了4个吸热峰,187 ℃处的第一个吸热峰对应着NaAlH4的融化过程,289、300和365 ℃3个吸热峰分别对应着NaAlH4的三步放氢过程,与文献报道的结果一致[19]。相比于NaAlH4,NaAlH4-6%TiB2@C三步放氢过程的吸热峰分别降低至156、202和337 ℃。此外,样品中NaAlH4的融化吸热峰消失,可能是由于TiB2@C的添加使得NaAlH4的分解放氢发生在融化之前造成的[20]。图6中MS曲线可知,TiB2@C的添加有效地降低了NaAlH4的放氢温度。

图6 NaAlH4-6%TiB2@C和NaAlH4的TGA、DSC及MS曲线图
图7为NaAlH4-6%TiB2@C和NaAlH4在常压下的等温放氢及速率曲线。由图7a可知,NaAlH4在160 ℃、常压下几乎不放氢,而NaAlH4-6%TiB2@C样品在100 ℃下180 min内的放氢量达到了1.4%,可见TiB2@C显著提高了NaAlH4的放氢动力学性能。随着温度的升高,NaAlH4-6%TiB2@C的放氢量和放氢速率明显上升。温度达到160 ℃时,样品在20 min内的放氢量为2.6%,在80 min内的放氢量达到了3.5%。由图7b可知,NaAlH4-6%TiB2@C在110~140 ℃之间40 min内可以完成放氢过程的80%,而温度上升到160 ℃时,完成时间则缩短至20 min。
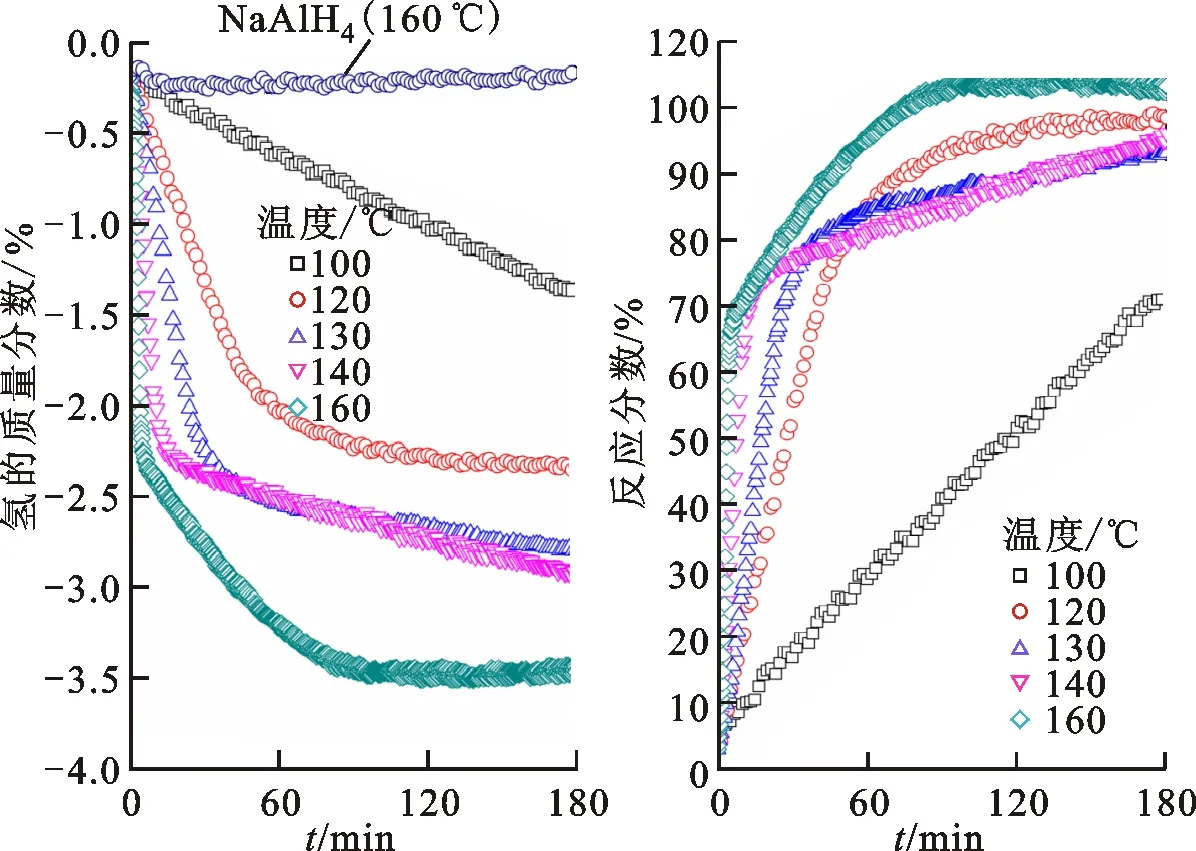
(a)等温放氢(b)速率曲线图7 NaAlH4-6%TiB2@C在常压下的测试曲线
为了比较NaAlH4和NaAlH4-6%TiB2@C的动力学性能,测试了两者在不同升温速率下(2~10 ℃/min)的TPD曲线,并采用Kissinger方法计算出两者前两步放氢过程的表观活化能(Ea),拟合公式如下
(1)
式中:Tm为TPD曲线的峰值温度;β是升温速率;R为气体常数;C为常数。TPD放氢曲线和Kissinger方法拟合曲线如图8所示。NaAlH4前两步放氢过程的表观活化能分别为165.5 kJ/mol和1 207 kJ/mol,添加TiB2@C后两者的Ea分别降低至96.5 kJ/mol和926 kJ/mol,这比纯TiB2催化的NaAlH4体系前两步放氢的表观活化能分别降低了9.4%和12.2%[14],比C60催化体系分别降低了11.5%和24.1%[21]。这些结果表明,TiB2@C复合催化剂较单一催化剂对NaAlH4和Na3AlH6放氢过程的催化效果更优,这是由于TiB2与C的协同催化,有效地促进H2的解离,从而有效地降低放氢能垒[22]。根据上述分析,在NaAlH4-6% TiB2@C中,一方面,TiB2均匀分布在颗粒表面能够起到活性催化位点的作用,促进H2的解离和键合,在放氢过程中促进[AlH4]和[AlH6]3-的分解,在吸氢过程中又能够促进NaH和Al的原位氢化形成NaAlH4[23];另一方面,C能够提高NaAlH4中Na与[AlH4]之间的电荷转移能力[24],从而削弱Al-H键,促进H2的解离与吸收,实现吸放氢可逆[24]。因此,TiB2与C的协同催化作用提高了体系的吸放氢动力学性能。
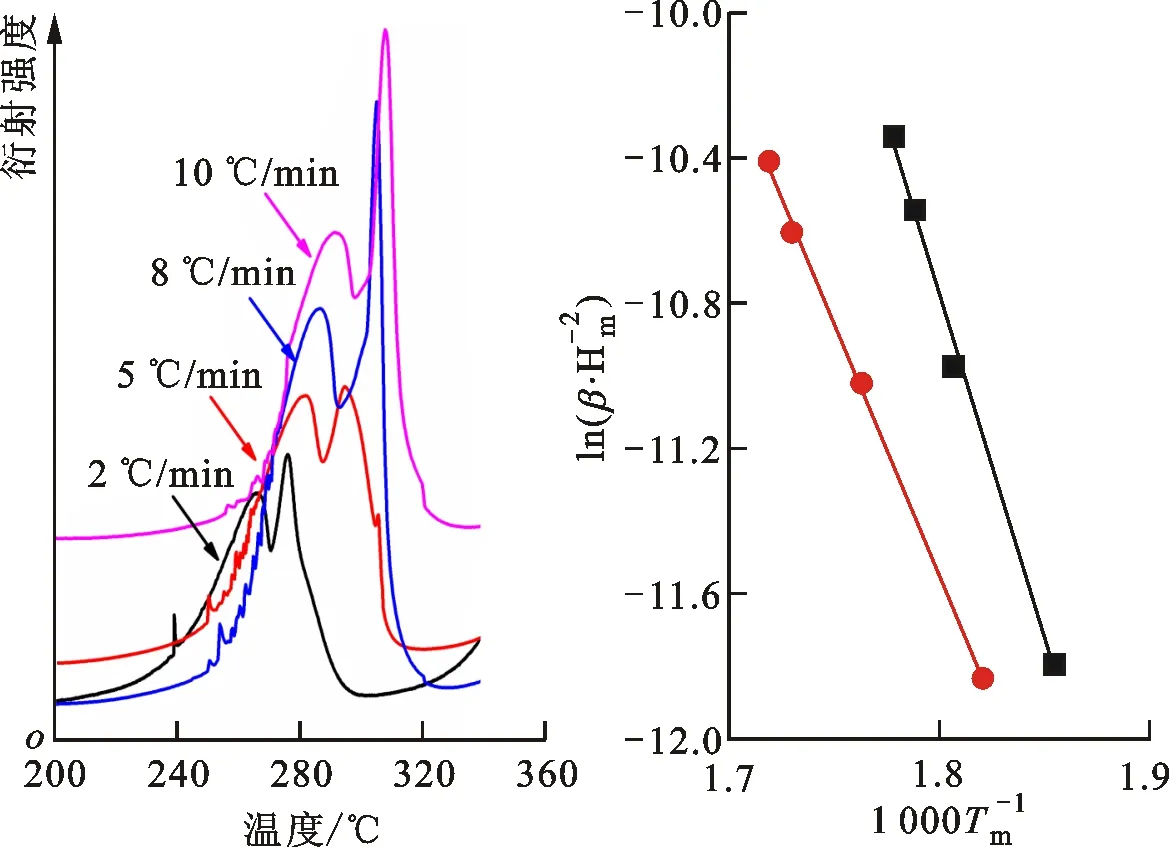
(a)NaAlH4

(b)NaAlH4-6%TiB2@C图8 NaAlH4和NaAlH4-6%TiB2@C在不同升温速率下的TPD放氢曲线和Kissinger法拟合曲线
为了考察NaAlH4-6%TiB2@C的可逆吸放氢性能,将NaAlH4-6%TiB2@C在40~240 ℃进行变温脱氢,然后在200 ℃、5 MPa氢压的条件下等温可逆吸氢,此过程重复10次。图9a为NaAlH4-6%TiB2@C在不同循环次数下的变温放氢曲线。NaAlH4-6%TiB2@C在10次循环过程中的可逆储氢量高达4.8%,并且由图9b可知,10次循环后吸氢产物均转变生成了NaAlH4,证明NaAlH4-6%TiB2@C具有优异的循环稳定性。值得注意是,图9a中从第1次到第10次的可逆放氢量略微有所降低,这是由于图9b中部分Na3AlH6和Al在可逆吸氢过程中并未完全转变成NaAlH4导致的。
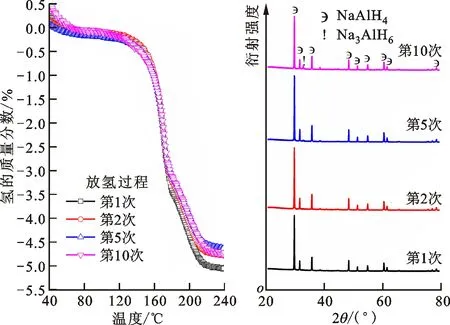
(a)不同循环次数下的(b)对应吸氢产物放氢曲线的XRD图谱图9 NaAlH4-6%TiB2@C的测试曲线
3 结 论
本文通过球磨和烧结的方法合成了非晶态TiB2@C催化剂,并在室温、5 MPa氢压的条件下球磨NaH和Al成功制备出NaAlH4。相比于球磨的纯NaAlH4,合成的NaAlH4-6%TiB2@C复合物具有更加优异的吸放氢动力学性能:起始放氢温度降低至10 ℃,三步放氢过程的峰值温度分别从289 ℃、300 ℃和365 ℃降低至156 ℃、202 ℃和337 ℃,前两步放氢过程的表观活化能分别从165.5 kJ/mol和1 207 kJ/mol降低至96.5 kJ/mol和96.2 kJ/mol,并且复合体系在160 ℃、常压下的放氢量达到了3.5%。此外,NaAlH4-6%TiB2@C还表现出优异的循环稳定性,在10次吸氢循环中的可逆储氢量高达4.8%。此性能的提高归结于TiB2@C中TiB2和C的协同催化效应,能够有效地促进吸放氢过程中H2的解离和键合,从而改善NaAlH4的吸放氢动力学性能。
猜你喜欢
分子催化(2022年1期)2022-11-02
油气与新能源(2022年4期)2022-08-30
汽车实用技术(2022年10期)2022-06-09
建材发展导向(2021年16期)2021-10-12
汽车文摘(2021年6期)2021-06-02
大自然探索(2021年11期)2021-01-05
智富时代(2018年3期)2018-06-11
智富时代(2018年3期)2018-06-11
中学化学(2016年10期)2017-01-07
Coco薇(2016年2期)2016-03-22
