
一步式光催化全水分解的研究进展
2021-01-30 05:31尹兴良李蕾蕾韩淑蕊
聊城大学学报(自然科学版) 2021年2期
尹兴良,李蕾蕾,韩淑蕊
(聊城大学 化学化工学院、山东省化学储能与新型电池技术重点实验室,山东 聊城 252059)
0 引言
光合作用是维持碳氧平衡和生态系统稳定的基础,其本质是一种天然的光催化过程。从1915年威尔施泰特首次从植物叶片中纯化出叶绿素起,科学家就一直试图通过某种技术实现人工光催化,但直到上世纪七十年代初本多-藤岛效应提出后,人工光催化才得以付诸实践[1-4]。近五十年来光催化研究取得长足发展,研究涉及水分解、污染物降解,氨合成,二氧化碳还原和有机物合成等多个领域[5-9]。其中光催化分解水以H2为能量载体对于实现“阳光能源计划”和缓解能源危机有潜在价值。光催化分解水有三种技术形式:(1) 光伏辅助电解水(PV-E);(2) 光电催化分解水(PEC);(3) 粉末光催化分解水。PV-E虽然效率较高,但其技术成本远高于已工业化的CH4-H2O重整产氢技术成本,因此不具有竞争力,而PEC和粉末光催化分解水的成本是4.1-10.4/kg和1.6-3.2/kg[10]。根据美国能源部计算的具有竞争力的H2价格是2.0-4.0/kg,以此区间为衡量标准,粉末光催化产氢更具有成本优势[11]。但值得一提的是光催化技术的成本计算的两个前提条件是光催化剂稳定运行10年以上和光转化为H2(STH)的效率10%以上,但至今还没有能同时满足两个前提条件的催化剂。纵观光催化的研究进展,研究者多关注有牺牲剂参与的光催化半反应而光催化纯水分解的研究则较少。但从未来光解水工业化需求和环境相容性方面考虑,后者更具有研究意义。
在全水分解研究中,有两类催化剂:一种是模拟植物光合作用两步光激发机理开发的由产氢、产氧半反应催化剂通过氧化还原电对结合构筑的两步式Z型结构催化剂;另一种是一步式光催化剂。Z型结构催化剂由于电荷转移效率较低和竞争反应,其表观量子产率可能存在上限[12],从所报道的文献看效率普遍偏低。有关Z型结构催化剂全水分解的综述文献较多,这些文献对该类型催化剂的叙述已较为系统全面。鉴于此,本文将集中介绍和讨论一步式全水分解光催化剂。本文首先介绍全水分解的机理,然后综述发展一步式光催化剂的策略和近期研究进展,最后概括分析一步式全水分解存在的问题和展望该类催化剂的发展方向。
1 全水分解机理分析
光催化全水分解是一吸热反应(ΔG=237 kJ/mol),因此需外加能量辅助。无催化条件下O-H键断裂需500 kJ/mol的能量,该能值远高于H2蕴含能值(284 kJ/mol),因此要实现全水分解的经济运行必须有催化剂的参与。光催化全水分解机理如图1(a)所示,其大致可以分为三个过程:(1) 催化剂受光激发产生电子-空穴对;(2) 电子-空穴对分离并各自迁移到催化剂表面;(3) 电子与质子结合产生H2,而空穴与OH-结合产生O2。上述是光催化反应的理想机理,实际催化过程中由于催化剂体相和体表存在缺陷,致使光生电荷复合而量子效率较低,因此开发光催化剂必须尽力降低电荷复合几率。另一方面光能的吸收利用也很重要,太阳光属复合光,一个太阳(AM 1.5)的标准光谱,可大致分为紫外光,可见光和红外光等三个区,所占能量的比例分别为5%,43%和52%[13],从光谱能量分布看,提高催化效率应开发一些对长波段光(如λ>400 nm)响应的催化剂。催化剂实现光催化全水分解的前提是催化剂导带(CB)和价带(VB)(对半导体类催化剂而言)或最高占有分子轨道(HOMO)和最低分子空轨道(LUMO)(对共轭高聚物类催化剂而言)要跨越产氢和产氧电位。如图1(b)所示标准H+/H2电位为0而OH-/O2电位为1.23 V,理想催化剂带宽为1.23 eV,但如果考虑过电位,则催化剂的带宽要大于1.23 eV,产氢和产氧基本电位需进一步拉伸到-0.41和1.6 eV。
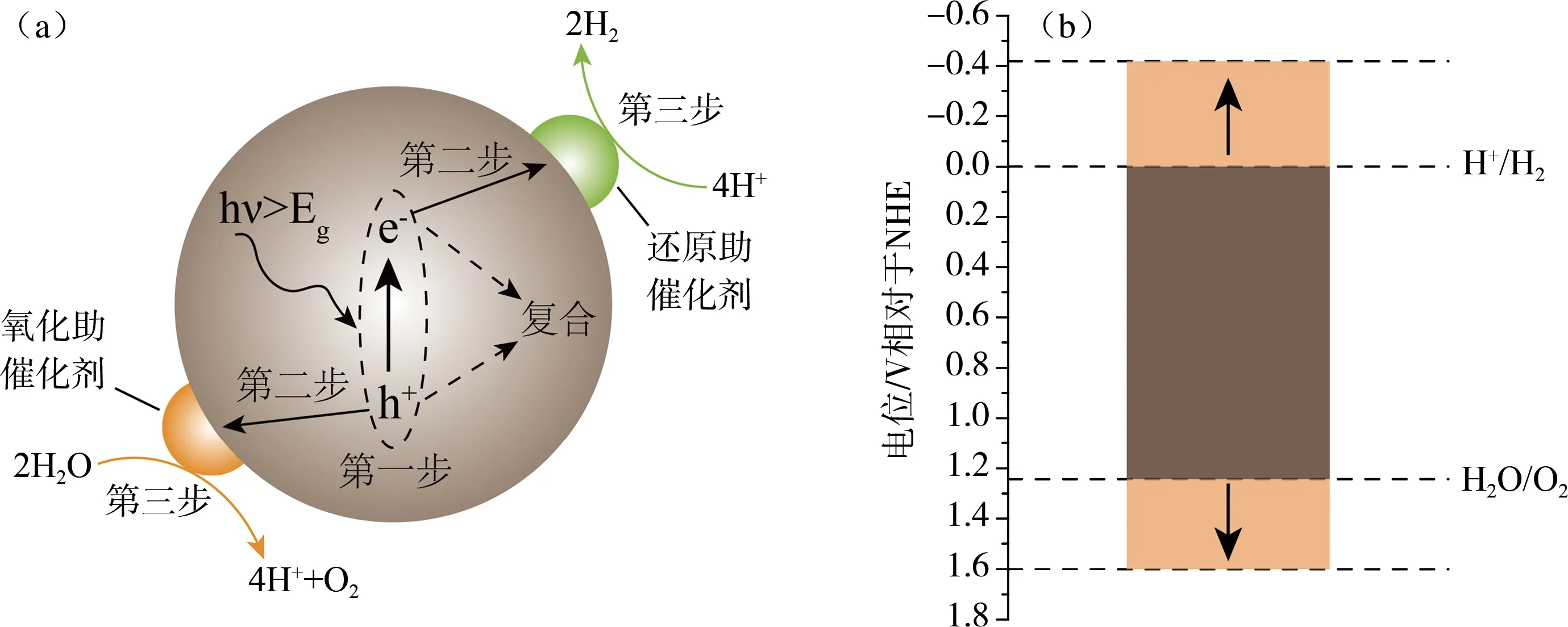
图1 (a) 光催化全水分解机理示意图;(b) 光催化全水分解氧化还原电位
2 光催化全水分解的策略及研究进展
由于宽带隙半导体(Eg> 3.0 eV)对光利用的局限,当前有关全水分解的研究多集中在具有可见光催化活性的催化剂上,一些具有里程碑意义研究工作的时间节点如图2所示。在这些工作的启发下,可见光驱动的全水分解得以进一步深入发展,近年来多有相关研究报道(表1)。为了更好地梳理研究成果,下文以构筑策略为线索综述了近年在一步式全水分解研究领域取得的进展。
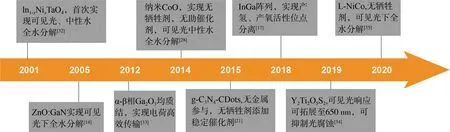
图2 一步式可见光催化剂重要研究时间节点
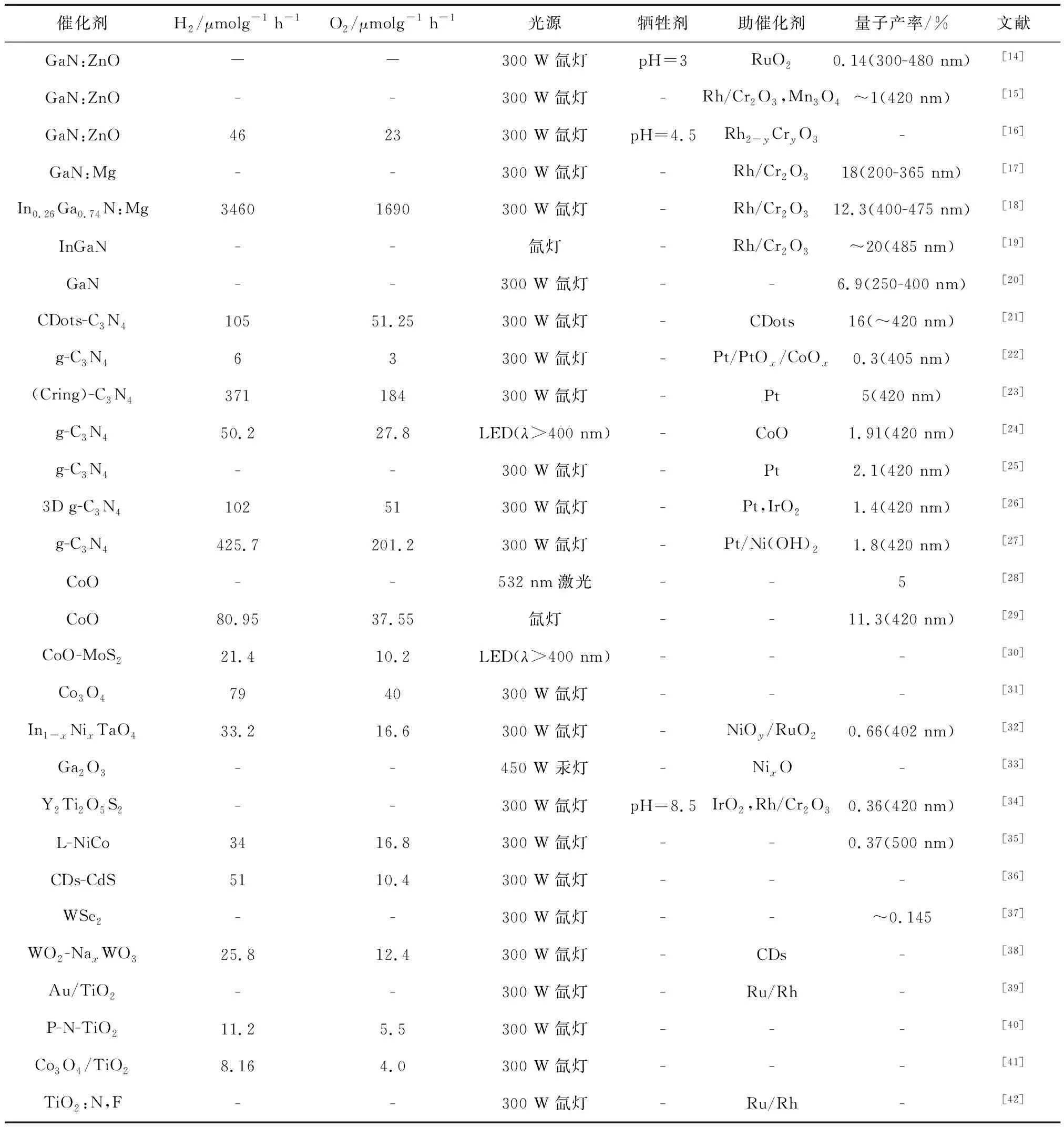
表1 近年具有可见光响应的典型全水分解催化剂
2.1 尺寸调控法
具有光催化全水分解活性的本体材料还未见报道,原因是本体材料的能带结构并不能够跨越产氢、产氧电位。量子限域效应在重构材料能带结构中发挥着重要的作用,将材料任一维度的尺寸降至激子波尔半径后材料的能级间隔增大,能带位置也会有一定移动,利用该效应可实现调控本体材料使之满足全水分解对电位的需求。最近这一策略应用在CoO、Co3O4、BiVO4和二维材料如黑磷,WSe2等本体材料上,制取了有催化全水分解性能的催化剂。2013年Bao课题组以此策略开发了具有分解中性水活性的纳米CoO,他们首先采用煅烧法将Co3O4相变为CoO,然后采用激光烧蚀和球磨技术将CoO粉末制备成d<10 nm的纳米颗粒。光照下所得纳米颗粒在无助催化剂,无牺牲剂的纯水中即可实现接近2:1计量比产氢,产氧的目标,而对比试验表明粉体CoO没有催化光解水活性。粉体CoO和纳米CoO的带边位置如图3(a)所示,粉体CoO的导带边位于产氢位更正的位置所以无法实现全水分解,而经尺寸调控所得的CoO纳米颗粒可实现导带和价带整体拉升,并跨越水氧化还原电位,因此可以实现全水分解[28]。受该工作启发翌年Guo课题组采用微波合成法合成了具有可见光全水分解催化活性的Co3O4-QDs量子点(d:3-4 nm),但是作为对比的固相法合成的Co3O4-SSR(20-60 nm)则没有活性。Co3O4-QDs和Co3O4-SSR能带结构如图3(b)所示,在可见光(λ>420 nm)(2.95 eV)照射下,虽然两者都不会发生Eg3(3.10-3.26 eV)跃迁,但是均可发生Eg1和Eg2跃迁,而对于Co3O4-SSR来说由于Co2+t2g上的电子还原能力不足而不能参与H+还原析氢半反应,而对于Co3O4-QDs由于量子效应的拉升作用,Co2+t2g上的电子还原能力高于H+/H2还原电势,因此Co3O4-QDs不但可氧化产氧还可以还原产氢,且两者比例接近1:2[31]。BiVO4作为可见光驱动的具有优异产氧性能的催化剂备受关注,块体BiVO4由于导带底趋向于0 V,使得光生电子能量不足以还原H+产氢。2014年Wang课题组采用水热法合成了BiVO4量子点,借助量子限域效应实现了导带提升,其提升后的导带底能位(ECB)小于pH=7的产氢电位-0.41 eV(见图3(c)),因此可以实现同步产H2和O2,但是该BiVO4量子点纯水中的产O2量高于产H2量,这里面的原因值得深入探究[43]。尽管如此,该工作实现了在无牺牲剂,无助催化剂条件下的中性全水分解,其意义仍然重大。二维材料是近年研究的热门材料,有多领域的广泛应用。在催化应用中,降低二维材料厚度不仅可以实现比表面积增加,优化传质,而且还可以实现能带调整,有望在全水分解中得以应用。具有半导体特性和各向异性的二维层状结构黑磷在催化、储能、光电和生物医学等方面有广阔的应用前景。传统的制备黑磷方法是在200 ℃,1.2 GPa高压下合成的,而制备纳米片层黑磷则需要用机械或者液相法剥离所得块体黑磷,上述获取纳米黑磷的方法耗时、耗能、价格昂贵,而且所得黑磷纳米片稳定性差。为了克服这一制约黑磷应用的瓶颈,Tian等人以白磷和乙二胺为原料采用溶剂热法一步合成了只有2-28原子层厚的黑磷,该法获得黑磷价格低廉,约1/kg相当于传统方法合成黑磷价格的1/500。更有趣的是所得黑磷可以实现长周期稳定催化中性水分解,λ> 600 nm时仍可以检测到H2,λ=420 nm所得最高量子产率~4%[44]。单层WSe2的带宽约为1.65 eV,吸光波长可以拓展到可见光区,带边结构跨越水氧化还原电位,具有催化全水分解的潜力。鉴于此Mi课题组采用分子束外延生长法在SiOx/Si基底上制备了单层WSe2,对比研究了双层和三层WSe2的中性水分解活性,结果表明仅有单层WSe2可催化全水分解,作者推测可能是由于双层和三层WSe2与水分解的氧化还原电位不相匹配所致。尽管这一推断需要进一步结合带隙结构分析验证,但是该结果证实了量子限域效应对材料带隙调整的有效性。单层WSe2催化活性评价结果表明所得H2/O2接近理论比值在1.95-2.1之间,STH约为0.145%,稳定运行8 h没有明显的活性变化[37]。除了WSe2外,计算表明一些具有垂直结构电场的单层二维材料如Al2Te3,Ga2Se3,Ga2Te3,In2S3,In2Se3和In2Te3等也有望成为全水分解的优异光催化剂,而且理论STH值均超过10%。需要特别强调的是对近红外光有吸收的In2Te3的理论STH值甚至高达32.1%[45]。同样的,Chen课题组计算结果表明单层PdSeO3也是一可见光下有望实现高效全水分解的催化剂,无助催化剂和牺牲剂条件下即可实现2:1 H2/O2产气比[46]。但到目前为止仍未开发出制备这些单层材料的工艺,因此对该类材料的研究今后应重点放在开发简单而廉价的制备方法上。
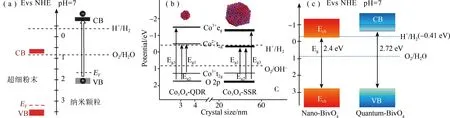
图3 (a) 粉体CoO和纳米CoO的能带结构[28];(b) Co3O4-QDs和Co3O4-SSR的能带结构[31];(c) 纳米BiVO4和量子点BiVO4的能带结构[43]
2.2 固溶体法
固溶体法是借助不同带隙半导体,通过合理调控比例实现带隙和带边调整以满足全水分解对能位的要求的一种方法。有关固溶体光催化剂具有里程碑意义的是Maeda等人报道的(Ga1-xZnx)(N1-xOx)。该课题组做了大量细致而深入的工作,2005年他们第一次报道了GaN-ZnO构筑的固溶体(Ga1-xZnx)(N1-xOx)在可见光下对全水分解有催化活性,但构成固溶体的GaN和ZnO均对全水分解没有活性[14]。ZnO和GaN属宽带隙半导体,带宽分别是3.2和3.4 eV,由于两者同属纤锌矿且有相似的晶格参数(GaN:a=b=0.319,c=0.519 nm; ZnO:a=b=0.325,c=0.521 nm),因此两者更易形成固溶体结构。有趣的是两者形成固溶体后带宽变窄并对可见光有响应,经密度泛函计算表明(Ga1-xZnx)(N1-xOx)固溶体的导带底主要由Ga的4s和4p轨道组成,而价带顶由N2p轨道和Zn3d轨道组成,较高价带中的N2p和Zn3d电子为价带最大值提供了p-d斥力,从而导致带隙变窄,吸收光谱红移(见图4)。尽管该固溶体吸光范围拓宽但催化全水分解效率较低,为提高活性该课题组详细研究了原料配比、氮化时间、助催化剂等对(Ga1-xZnx)(N1-xOx)活性的影响。结果表明x=0.12时,氮化15 h得到的纯相(Ga1-xZnx)(N1-xOx)活性最高[47],Ru2O作为助催化剂可以明显提高(Ga1-xZnx)(N1-xOx)的催化活性,但该体系对pH有一定的依存关系,pH=3时活性最高,铑铬氧化物较Ru2O的助催化活性提高了5倍多,但单独的铑氧化物或者铬氧化物并没有助催化活性,这一有趣实验现象文中缺乏深入讨论,如果加以研究或许对理解助催化剂的协同效应有帮助。此外研究还发现助催化剂不但有助于(Ga1-xZnx)(N1-xOx)固溶体催化效率的提升而且有助于维持催化剂的稳定性。以Rh2-yCryO3为产H2助催化剂,可维持催化剂稳定运行3个月,H2/O2比为2:1,6个月活性下降50%后,重新负载Rh2-yCryO3可使催化剂再生并可恢复至其初始活性的80%,进一步研究发现如果同时负载产O2助催化剂可以抑制(Ga1-xZnx)(N1-xOx)氧化分解,对维持稳定性有益[16]。影响(Ga1-xZnx)(N1-xOx)活性的因素除了e--h+复合外,还有水分解逆向反应,该课题组研究发现在长于(Ga1-xZnx)(N1-xOx)表面的Rh纳米颗粒外沉积镧系元素氧化物可抑制光解水逆向反应,提高催化活性。近来也有通过调控晶面和发展新型助催化剂如Rh2-yCryO3等提高(Ga1-xZnx)(N1-xOx)固溶体活性报道[16]。除了固溶体(Ga1-xZnx)(N1-xOx)可见光下对全水分解有活性外,Bi2Ga4O9、BiYWO6、(Zn1+xGe)(N2Ox)、In-Ni-Ta-O-N、AgTa1-xNbxO3、Bi0.5Dy0.5VO4等在可见光下也有催化全水分解活性,但是这些固溶体中除了In-Ni-Ta-O-N可以实现在无助催化剂条件下全水分解外,其余催化剂均需助催化剂辅助进行反应。
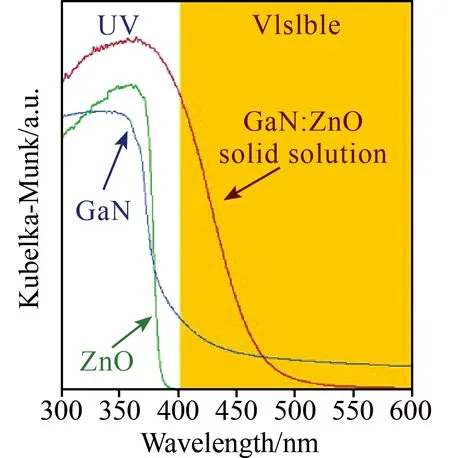
图4 GaN,ZnO和GaN:ZnO固溶体的紫外可见吸收光谱[14]
2.3 掺杂
全水分解早期(1980-2000年)研究多集中在一些宽带隙半导体催化剂上,二十年间开发了几百种氧化物催化剂,如NaTaO3、K4Nb6O17、ZnGa2O4等,但是这些催化剂仅能吸收利用太阳光谱中的紫外光,如上所述紫外光在光谱中能量占比较小,因此光能的利用率比较低,这导致量子效率不高。宽带隙半导体窄化利用是一提高其效率的策略,应用比较多的是掺杂法,通过引入杂原子实现带隙的窄化。如图5是阳离子和阴离子掺杂的示意图,对于阳离子掺杂,引入的阳离子会在宽带隙催化剂中产生杂质能级,或在原半导体的价带以上产生新的施主能级,或在原半导体的导带以下产生新的受主能级,从而起到窄化带隙的目的。而对于阴离子掺杂,借助掺入的阴离子与半导体中阴离子杂化后将原价带电位提升形成新的价带从而起到窄化的目的。如图6(a)所示,AgTaO3(Eg~3.4 eV)属宽带隙半导体,仅能在紫外光下催化全水分解,其导带和价带电位分别约为-0.9和2.5 V,而AgNbO3仅能作为半反应催化剂,其导带和价带分别约为-0.3和2.5 V。两者相比AgNbO3的导带更正,而两者的导带分别由Ta 5d轨道和Nb 4d轨道组成,如果由Nb部分取代Ta可能会使AgTaO3的带隙窄化从而可以吸收可见光分解水。基于此考虑,Irie课题组制备了AgTa1-xNbxO3并考察了掺杂比例对活性的影响,最终发现NiO/AgTa1-xNbxO3可实现H2/O2比2:1,并可实现192 h长周期稳定运行[48]。可见光照射下(λ> 420 nm) InTaO4本身有催化全水分解的活性,但是活性较低,通过Ni掺杂提升InTaO4价带电位可窄化其带宽(2.6→2.3 eV),拓展光吸收范围(420→520 nm),更利于氧化物电子由价带激发到导带上进而有利于提高催化活性。稳定性测试表明催化剂运行400 h未见活性降低,所得H2/O2接近2,在λ=420 nm处量子产率为0.66%。尽管该工作所得In1-xNixTaO4(x=0-0.2)的活性较低,制备过程耗能高(1100 ℃)且需要NiOx作为助催化剂,但该研究为后续的全水分解催化剂的开发提供了思路[32]。掺杂除了可以调控半导体带隙还可以促进电荷传递和提高催化剂的稳定性,第III主族的氮化物如Ga(In)N带隙跨越全水分解的电位要求,吸光范围从紫外光到近红外光,研究发现提高材料中N含量可以抑制光腐蚀和氧化。Chowdhury等采用等离子体辅助分子束外延生长法制备了In0.22Ga0.78N纳米片阵列(图6(b)),并控制Al掺杂在纳米片的一个面上构筑了内建电场(图6(c)),实现光激发电荷的有效分离和定向传输,并且实现了氧化还原电位的空间分离从而抑制了水分解逆向反应[19]。虽然掺杂对催化效率的提升有积极的促进作用,但是需要注意的是杂原子的掺入也有影响效率提升的负面效应。掺杂通常会引入不连续能级,因此会减慢光生电荷的迁移速率。而且引入杂原子很难保持电荷的平衡,这会导致作为复合中心的空位产生,从而增加e--h+复合[49]。
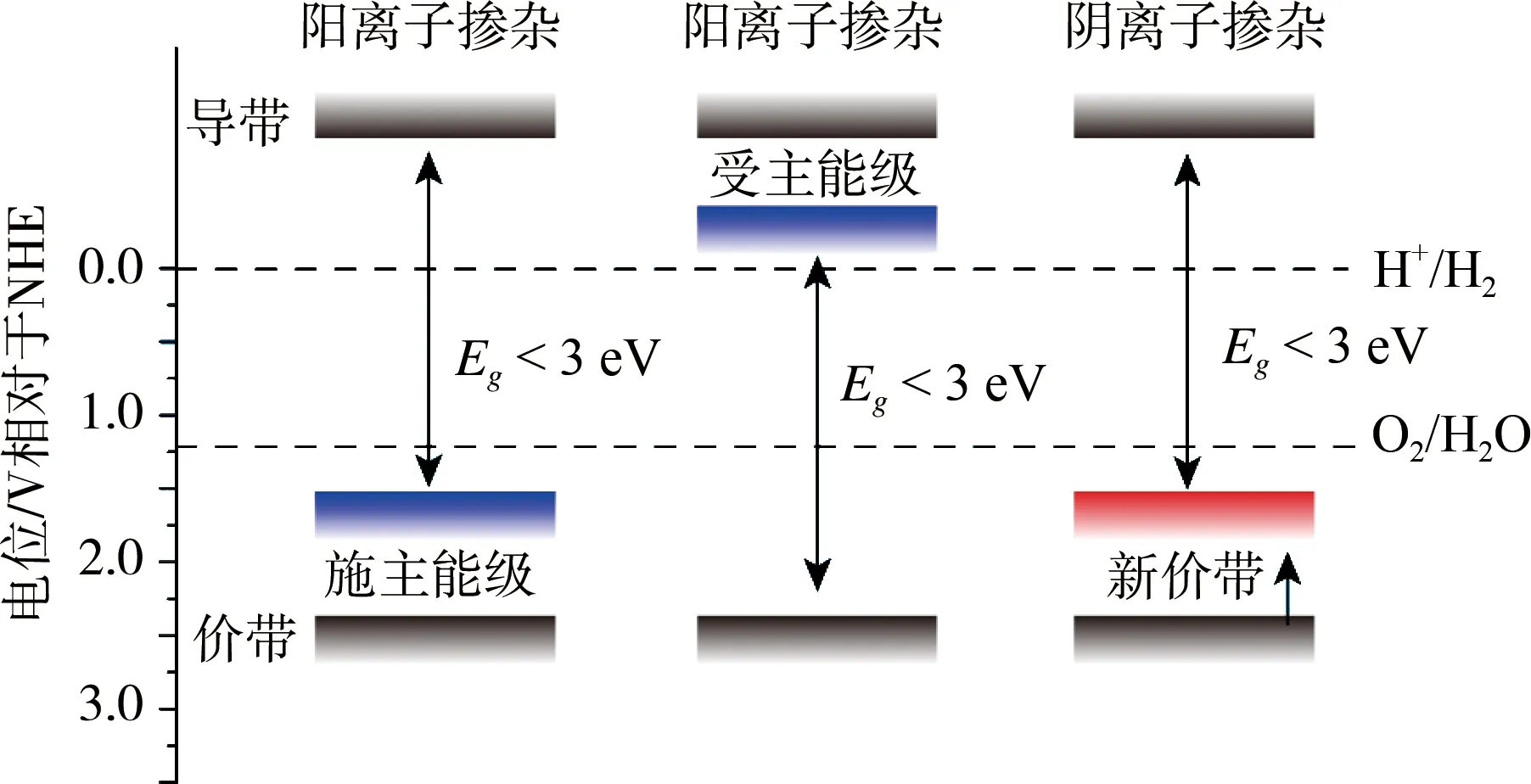
图5 阴、阳离子掺杂窄化带隙示意图

图6 (a) AgTaO3、AgNbO3和AgTa1-xNbxO3的能带结构[48];(b) InGaN:Mg的SEM图像;(c) InGaN:Mg的电荷传递示意图[19]
2.4 表界面调控
光催化全水分解反应在催化剂的表界面进行,过程受表面电荷特性、能带结构、反应产物等因素影响,因此对表界面深入研究有助于指导高效催化剂的合成。GaN能带结构满足H2O分解电位需求,但GaN表面能带弯曲增加了电荷传输至催化剂与水的界面处能耗,进而降低了反应速率,而合成具有可控表面电荷特性(可调节的表面费米能级和能带弯曲)的纳米结构GaN一直是一挑战。Kibria等通过Mg掺杂GaN表面实现了对GaN表面费米能级调控,通过平衡氧化/还原反应以达到提升催化效率和稳定性的目的[17]。进一步将该策略施行于InGaN中该课题组获得了双P型GaN/In0.2Ga0.8N其量子产率高达~12.3%, 该值是目前报道的中性全水分解最高值[18],但是该催化体系仍需贵金属助催化剂的辅助,如果发展可替代的廉价助催化剂或许能保持高量子产率的前提下降低催化剂制备成本。催化剂表面的一些不纯相也会影响催化反应进行,如前所述BiVO4是一类性能优异的产氧光催化剂,由于其固有的导带电位高于氢还原电位,因此难以实现产氢。通过V掺杂和Bi原子部分取代而合成固溶体BixM1-xVO4, BiVO4导带边得以抬升而可实现同时产H2和O2。但最近Fang等发现不同批次合成的BixY1-xVO4的颜色和活性不同,尽管所合成的BixY1-xVO4在可见光下有吸收但并不能在可见光下实现全水分解。研究发现颜色不同和吸收拖尾(~500 nm)与BixY1-xVO4表面的BiOy团簇有关,BiOy团簇和BixY1-xVO4可形成异质结,但由于其导带边较产氢电位更低,因此由BiOy通过异质结捕获的电子并不能实现产氢。基于此研究发现,他们采用稀酸(HNO3)浸渍除去表面的BiOx团簇,之后所得BixY1-xVO4的全水分解活性可提升4倍[50]。石墨状C3N4(g-C3N4)是一类廉价易得、环境友好、具有可见光响应的催化剂,但由于纯相g-C3N4催化过程中产生的H2O2的毒化作用使得g-C3N4催化水分解必须在牺牲剂存在的条件下才能实现。尽管搅拌、鼓泡或加入化学试剂等方法试图克服这一缺陷,但收效甚微。Kang课题组利用碳量子(CDots)点修饰g-C3N4表面,借助CDots 独特的催化H2O2分解特性有效克服了上述不足,实现了在无牺牲剂参与下长周期(一年)稳定分解水产H2/O2的目的,所得H2和O2比为2:1[21]。根据该工作的原理,该课题组又发展了一系列全水分解的催化剂如CDots-CdS,CoO-g-C3N4,WO2-NaxWO3-CDots等,同样取得不错的全水分解活性和稳定性[24,38,51]。TiO2是最早研究的一类环境相容性好,稳定性高的光催化材料,有锐钛矿、金红石和板钛矿三种晶型,其中仅有金红石相有全水分解活性,有研究发现锐钛矿TiO2通过Mg掺杂消除其缺陷态后也表现出全水分解的活性。过去有较多报道TiO2全水分解的文献,但多数是在紫外光或者全光谱下进行反应,光利用效率不高,因此拓展光响应范围成为TiO2全水分解研究的重点。掺杂和半导体敏化可以拓展TiO2的吸光范围,但正如前述掺杂引入的缺陷态可能会成为新的电荷复合中心,而CdS、CdSe等一些有毒半导体的引入会带来一些环境问题。鉴于此wang等发展了一种电沉积法,在TiO2纳米管阵列表面可控沉积了绿色价廉的CDots,所得催化剂的吸光范围可拓展至近红外光区[52]。该催化剂在可见光区表现出良好的污染物降解活性和全水分解活性。除CDots外,Co3O4也有很好的敏化作用,纯TiO2在可见光下没有活性,但催化剂Co3O4-TiO2在可见光照射下产氢、产氧活性分别为8.16和4.0 μmol g-1h-1,明显高于纯Co3O4的催化活性[41]。最近发展的利用表面等离激元共振效应调控表界面技术也有助于拓展TiO2等材料的光吸收[39,53]。从Moskovits等研究开始,Au-TiO2等离激元催化体系得到广泛研究,依靠金属的表面等离子体共振效应可大大提升催化剂光谱响应。2017年李灿课题组构筑了Au-TiO2肖特基结[39],在λ> 440 nm可见光照射下对比纯TiO2和Au-SiO2未表现出产氧活性,而Au-TiO2表现出很强的产氧活性。活性测试表明TiO2表面Au等离激元激发作用在促进水氧化中发挥重要作用,进一步研究发现富集于Au-TiO2界面处的等离子体空穴作为活性位点参与了水氧化过程。该研究揭示了等离激元催化剂在全水分解中的催化机理,对研究其它类似催化体系提供了借鉴(图7(a))。除TiO2外,等离激元效应应用于半导体CdS上同样可催化全水分解,如图7(b)所示,HCAu@#CdS/TiO2核壳结构等离激元催化剂在可见光照射下,由Au等离子体效应产生的热电子会注入到CdS的导带中进而与H+反应产生H2,而留在Au中的空穴与H2O发生反应产生O2,该催化体系不但提高了催化活性而且有效保护了CdS免于光腐蚀,催化全水分解稳定运行3 D而没有活性衰减[54]。
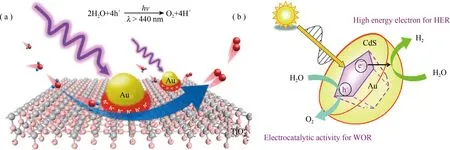
图7 (a) 等离基元光催化剂Au/TiO2可见光全水分解示意图[39];(b) 等离基元光催化剂Au@#CdS/TiO2催化全水分解示意图[54]
2.5 拓展π-π共轭
作为有机聚合物类光催化剂g-C3N4,虽然其N2p(HOMO)和C2p(LOMO)轨道跨越全水分解电位值,但由于g-C3N4表面缺少氧化还原活性位点,其固有高激子结合能导致光生电荷利用率低等缺陷使得其难以实现全水分解。通过构筑双助催化剂型催化体系如Pt-CoP-g-C3N4、Pt-PtOx-CoOx-g-C3N4可实现无牺牲剂中性全水分解,但这种有机-无机异质结界面弱相互作用和晶格、能带不匹配致使其结构不稳定和电荷迁移率低。近来,韦世强和刘庆华课题组首次在分子水平上构筑了面内全水分解异质结,他们以葡萄糖和三聚氰胺为前驱体采用共聚法构筑了C环- g-C3N4面内异质结,借助强sp2杂化电子结构在费米能级附近富有自由电子的特性实现带隙窄化和导带边正向移动,同时形成的微观π-π共轭界面电荷梯度可推动电子-空穴分离并反向传递(见图8)。这种π-π共轭异质结可显著改善 g-C3N4的吸光性能和反应动力学性能,相比于纯相g-C3N4光生载流子的扩散长度和寿命提高了10倍,实现了无牺牲剂、无助催化剂、中性条件下全水分解[23]。同样的Gangavarapu课题组实验也印证了拓展π-π共轭对提升g-C3N4活性和实现全水分解所发挥的积极作用。他们采用甲胺与TAB共聚的方法合成了苯环- g-C3N4催化剂,苯环的引入借助π-π共轭效应可以使g-C3N4的带隙由2.8降至2.1 eV。 DFT计算表明苯环掺杂后在庚嗪环不同部分的价带最大值和导带最小值的局部电荷密度有助于降低电荷复合[25]。最近Fang等采用共晶-热处理两步法制备了富碳g-C3N4(Cco-C3N4),他们首先以TPA和MA为原料,通过TPA上的-COOH与MA上的-NH2形成氢键构筑二维分子平面,TPA芳环引入有利于增强富碳骨架π-π共轭效应,以促进电荷的分离和传输,在纯水、无牺牲剂和助催化剂条件下实现产氢产氧分别为530和255 μmol g-1h-1,高于其它报道的非金属催化剂活性[55]。进一步对带隙分析,发现富碳单元的嵌入引入了新的中间能级,窄化了g-C3N4带隙使得吸光范围从460拓宽至800 nm,大大提高了光的利用率。除此之外,富碳单元与g-C3N4构筑的异质结对电荷分离有很大的促进作用,在上述两优势的影响下活性得以提升。除g-C3N4外一些具有全水分解活性的其它二维高聚物类催化剂也广受关注,由于二维聚合物独特的理化性质、光电性质、结构多样性和π-π共轭平面内电子高度离域化等有助于设计新型高效全水分解催化剂。从1985年第一次报道聚合物光催化剂至今,尽管在该领域取得了很大发展,但具有全水分解活性的由单一单体构筑的聚合物催化剂仍然比较少。通过构筑异质结有助于二维高聚物电荷的分离传输,wang等设计构筑了CMP/C2N二维范德华异质结,X射线近边吸收光谱表明CMP和C2N之间形成了紧密的异质结,由于优质异质结的形成,光生电荷复合得以抑制,该催化剂在600 nm处表观量子产率达到4.3%。
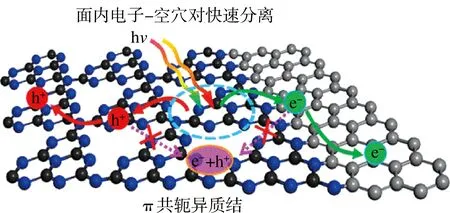
图8 π共轭面内异质结电荷分离迁移示意图[23]
2.6 形貌结构调控
g-C3N4是典型的二维结构材料,层与层间的堆叠易减少比表面积和降低光生载流子传输速率,如果能以2D g-C3N4为模块自组装成3D结构则有望解决上述弊端。鉴于此,Zhang课题组设计合成了一种高度多孔的超薄纳米片,通过自下而上的超分子自组装路线构筑了三维多孔石墨状氮化碳(3D g-C3N4NS)。制备的3D g-C3N4NS呈现3D互连开放框架结构,具有极大比表面积,可以有效抑制纳米片堆叠,改善载流子传质和光子吸收。在可见光下,3D g-C3N4NS同时实现了稳定,高效的光催化全水分解,在纯水中H2和O2的生成速率分别高达102和51 μmol g-1h-1[26]。但需要注意的是上述工作仍需要负载贵金属助催化剂,为了降低催化剂成本,Xiong等在缺陷3D g-C3N4上生长WC1-x构筑了WC1-x/g-C3N4催化剂,在无贵金属助催化剂、无牺牲剂、可见光条件下实现全水分解(H2:84.1 μmol h-1g-1; O2:41.7 μmol h-1g-1)[56]。除了增加催化剂的比表面积和活性位点外,还可以通过形貌调控抑制全水分解的逆向反应。如图9所示,一般全水分解产生的O2会逆向吸附到产氢活性位点上与H2反应生成水从而降低催化活性。研究发现如果在半导体催化剂表面光沉积ⅣⅤ族过渡金属(Ti,Nb,Ta)非晶态羟基氧化物形成核壳结构,借助羟基氧化物分子筛作用选择性过滤反应物和产物分子则可以有效抑制全水分解的逆向反应[57]。此外构筑核壳结构还有助于形成异质结和改善表面电荷反应动力学性能。Fu等采用溶胶凝胶法构筑了GaInZnON@GaInON I型核壳结构异质结,通过带边结构的匹配,光生电荷容易从GaInZnON核传递到GaInON壳上,而且DFT计算表明在近GaInON壳费米能级处形成的中间能级更利于空穴的传输。由于上述两个优势该催化剂表现出良好的催化活性,430 nm光照下量子产率为3.5%[58]。类似地, Zhang等以Co-Zn双金属MOF为前驱体构筑了多孔Co3O4/ZnO@ZnS核壳结构异质结。该结构中壳状Co3O4附着于ZnO@ZnS外,相比于不连续分布的Co3O4更能抑制表面电荷复合。由于ZnO@ZnS异质结形成的内建电场和Co3O4对表面电荷反应动力学的改善,电荷传递效率和反应速率得以提升[59]。
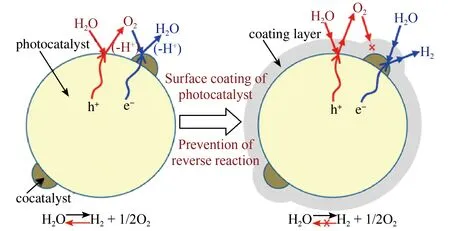
图9 核壳结构抑制光催化全水分解逆向反应机理示意图[57]
3 结论和展望
光解水是转化利用太阳能的绿色技术,一步式光解水催化剂制备简单、无需电解质和导电基底、无需施加外部偏压,因此更具有技术和成本优势。相对于有牺牲剂参与的光解水半反应而言,无牺牲剂参与的全水分解更有现实意义。本文以催化剂的构筑为切入点综述了近年有可见光响应的全水分解催化剂的研究进展。虽然该领域研究取得了很大的进步,但如下几个问题仍然有待进一步解决。
(1) 一步式全水分解催化剂的阴阳极集成于一体,催化产物为H2和O2混合气体,如果不能有效分离不但不能很好地加以利用还有潜在的安全问题,因此应发展高效分离H2和O2的技术。
(2) 一步式光催化产氢和产氧活性位点往往混杂在一起,易促进水分解的逆向反应,这也是导致全水分解效率低的重要原因,因此如何构建分离的氧化、还原活性位点尤为重要。尽管已有一些解决该问题的报道如表面助催化剂隔离法,保护活性位点法等,但发展更普适性的方法仍然面临很多挑战。除此之外,如何辨别产氢、产氧活性位点也面临很多困难。未来解决这些问题应该更多地借助一些先进的原位成像技术和瞬态技术。
(3) 尽管近年不乏可见光响应全水分解催化体系报道,但从光谱响应上看可见光区域的吸收比较弱,光能利用效率比较低,因此今后研究应该更加关注如何提高光能的利用。提高光的利用率应从两个方面展开研究:1) 满足全水分解电位要求的前提下,进一步窄化带隙,发展新型催化剂;2) 发展上转化材料,将难以利用的红外光转化为低波段的可见光加以利用。
猜你喜欢
家庭教育报·教师论坛(2021年42期)2021-12-23
疯狂英语·新阅版(2021年9期)2021-10-30
北方论丛(2021年2期)2021-05-22
现代装饰(2021年1期)2021-03-29
中学生数理化(高中版.高考理化)(2020年10期)2020-10-27
分析化学(2019年3期)2019-03-30
中国科技纵横(2019年3期)2019-03-25
分析化学(2017年12期)2017-12-25
新高考·高一物理(2016年7期)2017-01-23
新高考·高一物理(2015年6期)2015-09-28
