
南极菲尔德斯半岛潮间带沉积物细菌群落结构分析及产酶菌株初步筛选
2021-01-14 11:43吴蕾蕾商丽孙浩史晓翀张晓华
极地研究 2020年4期
吴蕾蕾 商丽 孙浩 史晓翀,2 张晓华,2
研究论文
南极菲尔德斯半岛潮间带沉积物细菌群落结构分析及产酶菌株初步筛选
吴蕾蕾1商丽1孙浩1史晓翀1,2张晓华1,2
(1中国海洋大学海洋生命学院, 山东 青岛 266003;2青岛海洋科学与技术试点国家实验室海洋生态与环境科学功能实验室, 山东 青岛 266071)
为了研究南极菲尔德斯半岛潮间带沉积物细菌群落组成及可培养细菌产酶活性, 选取潮间带19个站位沉积物样品, 利用Illumina MiSeq高通量测序技术对沉积物中细菌的群落组成进行研究; 利用荧光定量PCR技术定量测量沉积物中总细菌的绝对丰度; 利用传统的平板涂布法对16个站位的沉积物样品中可培养细菌进行纯化和鉴定, 同时选取38株细菌进行酶活性筛选。结果显示: 19个站位潮间带沉积物样品共得到2 375个分类操作单元OTU(operational taxonomic unit), 共获得42个细菌门, 其中变形菌门(Proteobacteria)、拟杆菌门(Bacteroidetes)、放线菌门(Actinobacteria)为优势类群。总细菌的丰度介于2.51×107~6.65×108copies·g–1。对于可培养细菌, 129株测序的细菌分属于4个门, 25个属, 50个种。变形菌门、放线菌门、拟杆菌门为优势类群, 这与高通量测序结果基本一致。选取其中38株进行酶活性筛选(包括脂酶、淀粉酶、明胶酶、褐藻胶酶和纤维素酶), 发现17株可产脂酶、24株可产淀粉酶、18株可产明胶酶、4株可产褐藻胶酶和4株可产纤维素酶。
菲尔德斯半岛潮间带沉积物 高通量测序 可培养细菌 16S rRNA基因 酶活性
0 引言
南极位于地球最南端, 全年冰雪覆盖, 酷寒干燥、强光辐射, 土壤养分贫乏, 为典型的极端环境[1]。南极地区没有大型植物以及大型食草类动物存在, 植物大多为地衣、苔藓, 同时微生物也成为优势物种[2]。南极微生物在极地生物地球化学循环中发挥重要作用, 并且在长期进化的过程中其生态特征及群落结构已经适应南极的极端环境[3]。因此研究该地区微生物多样性有助于加深对南极生物地球化学循环过程的理解, 探究微生物在南极地区生物地球化学循环过程中发挥的作用, 同时为开发南极可利用的微生物资源提供基础。
菲尔德斯半岛位于南设得兰群岛的乔治王岛西南端, 为无冰区, 属于亚南极地区, 并受到海洋气候的影响。在过去的50年期间, 多个国家在半岛建立科考站, 大多数人类活动及陆地生物研究也都聚集在该半岛。该半岛的西海岸是重要的海豹栖息地, 东海岸设有多国考察站, 动物及人类活动对该地区的环境造成了一定程度的影响。前期对菲尔德斯半岛细菌多样性进行了一些研究, Xiao等[4]利用变性梯度凝胶电泳(DGGE)的方式对长城站附近海水及土壤样品细菌多样性进行研究, 结果表明在土壤及海水样品中有丰度较高的γ-变形菌纲 (Gammaproteobacteria)、放线菌纲 (Actinobacteria)、黄杆菌纲(Flavobacteria)和厚壁菌门(Firmicutes)。Zeng等[5]利用454焦磷酸测序对阿德雷湾及长城湾附近表层海水样品微生物多样性进行分析, 发现阿德雷湾样品细菌可划分为18个门类, 长城湾样品细菌分属于11个门类, 其中拟杆菌门、变形菌门为优势细菌。在可培养方面, 杨晓等[6]对半岛土壤样品的细菌多样性进行研究, 并阐述了人类活动及企鹅、海豹对细菌多样性的影响。这些研究证实了尽管处于较为特殊的低温环境中, 菲尔德斯半岛的微生物群落仍有较高的丰度和多样性。此外, 还有研究对可培养菌株进行低温酶筛选, 获得了产蛋白酶、脂肪酶等活性的菌株[7-8], 用于后续低温酶的开发使用。然而这些研究仅通过非可培养或可培养单一方法阐释了半岛细菌群落组成, 通过高通量测序与传统平板培养法结合来研究潮间带沉积物环境样品中细菌的群落组成的研究目前还很少。
本研究将2016年中国第32次南极科学考察采集的菲尔德斯半岛潮间带19个站位的沉积物作为研究对象, 通过高通量测序分析此地区细菌群落结构, 利用荧光定量PCR测定环境中总细菌丰度。同时利用传统分离培养法对16个站位的沉积物样品可培养细菌进行分离鉴定, 初步揭示潮间带沉积物中可培养细菌的多样性。同时选取其中38株分离的菌株进行产酶活性研究, 为开发极地低温酶资源提供数据。
1 材料与方法
1.1 样品采集
本实验所用样品采自2015—2016年间南极菲尔德斯半岛表层0~2 cm潮间带沉积物。共获得19个沉积物样品, 其中西海岸7个样品(W1~W7), 东海岸12个样品(E1~E3, E5~E13)(图1, 表1)。用无菌铲铲取适量沉积物样品放入无菌自封袋中, 于-20℃冰箱保存, 返回实验室后保存在-80℃冰箱用于后续DNA提取。选取16个站位的沉积物样品(除E8、E11、E12), 称取约1 g于15 mL 0.85% (w/v)的生理盐水中混匀, 吸取100 μL于900 μL无菌生理盐水中混匀, 吸取100 μL的浑浊液现场涂布于2216E(MA)、R2A培养基[9]上, 4℃培养, 返回实验室进行分离和鉴定。
1.2 菌株DNA提取和高通量测序
取0.5 g沉积物(湿重), 使用E.Z.N.A土壤DNA取试剂盒(Omega, 美国)[10], 按照试剂盒操作说明进行总DNA提取并用凝胶电泳法和NanoDrop-2000分光光度计检测DNA的浓度及质量。选取16S rRNA基因V4区, 利用引物515F(5’-GTGCCAGCMGCCGCGGTAA-3’)和806R(5’- GGACTACHVGGGTWTCTAAT-3’)对16S rRNA基因片段进行PCR扩增[11], PCR产物用AxyPrepDNA凝胶回收试剂盒(Axygen, 美国)进行纯化与浓缩[12], 利用Illumina MiSeq平台进行高通量测序。其中PCR扩增、纯化和建库由上海美吉生物医药有限公司完成, 测序所得原始序列上传至NCBI数据库, 登录号为: SRP226771。
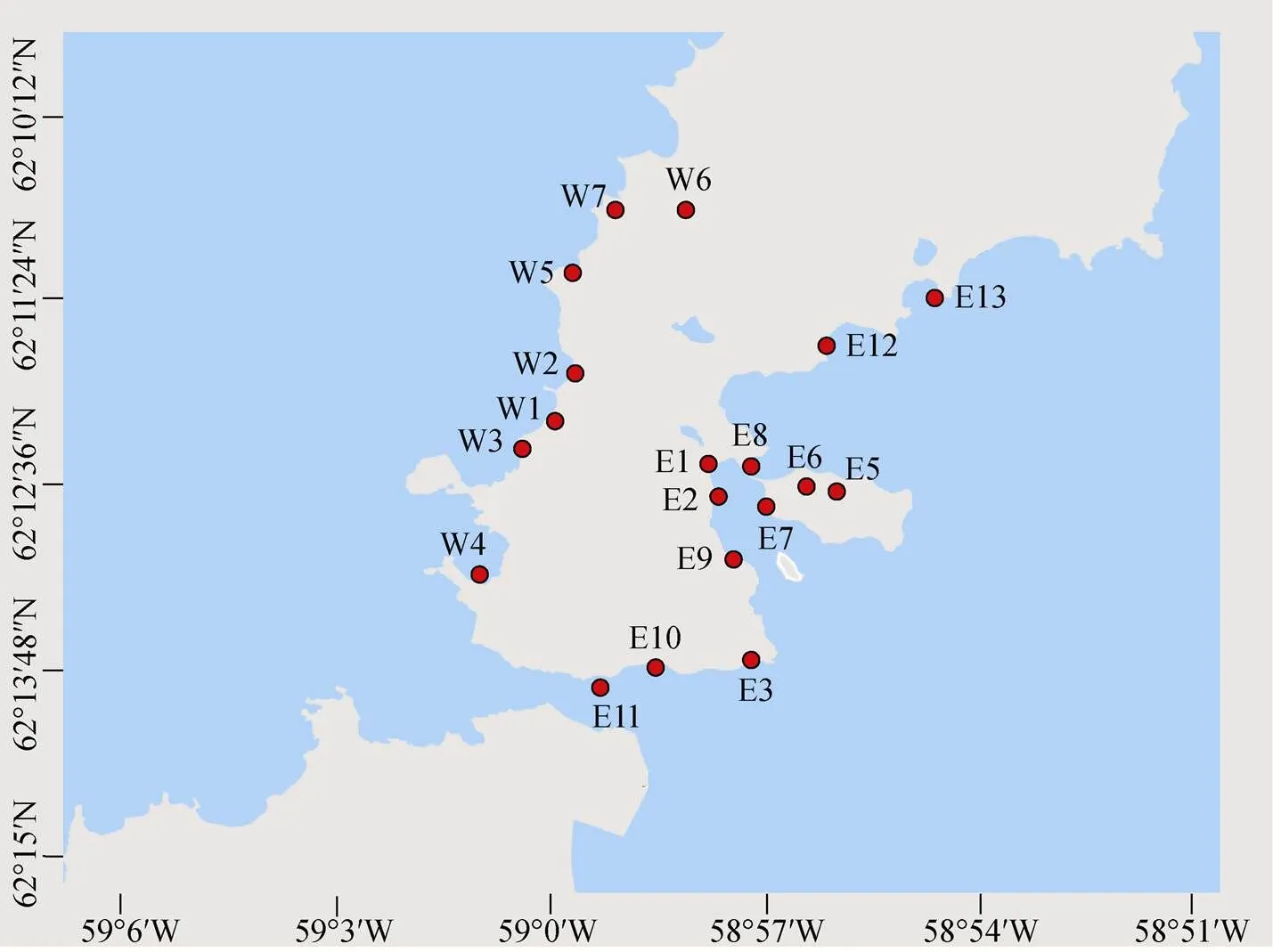
图1 菲尔德斯半岛潮间带沉积物样品采集站位图
Fig.1. Sampling sites of intertidal sediment samples in the Fildes Peninsula

表1 菲尔德斯半岛潮间带沉积物样品采集位点信息表
1.3 荧光定量PCR
本实验室采用荧光定量PCR的方法进行细菌丰度的绝对定量。采用细菌通用引物515F/806R扩增目的片段。具体试剂及操作见参考文献[13]。
1.4 数据处理
Illumina MiSeq测序得到的数据利用Mothur 软件[14]进行去杂得到有效的序列。利用Usearch平台将得到的有效序列进行OTU聚类, 并在97%的相似性水平下根据Silva数据库进行物种的分类注释。α多样性通常包含Shannon指数、Chao1指数和Good’s物种覆盖度等[15-17]。对于β多样性, 在OTU的分类水平上基于Bray-Curtis距离进行非度量多维尺度分析(non-metric multidimensional scaling, NMDS)用于揭示样品之间的聚簇, 并利用ANOSIM分析检验差异。利用Canoco 5.0软件进行db-RDA分析以揭示物种聚类及其对环境因子的响应情况。东西部样品物种分布的差异检验由IBM SPSS25.0完成, 利用非参数检验中的Mann-Whitney U检验方法进行计算, 同时利用相同方法与2012年本实验室的分析结果进行比较分析[18]。
1.5 可培养菌株的分离鉴定及酶活验证
根据颜色、大小、形态的不同挑取菌落, 三次划线分离纯化后利用酚氯仿法提取DNA[19]。利用细菌16S rRNA基因通用引物B8F/B1510进行PCR扩增[20]。PCR产物用限制性内切酶III酶切后进行 RFLP分析, 选取图谱不同的PCR产物送北京六合华大有限公司测序[21]。通过EzBioCloud数据库(https://www.ezbiocloud.net/)对测序结果进行比对, 确定菌株的分类地位[22]。对于淀粉酶、脂酶、明胶酶、褐藻胶酶及纤维素酶平板的配制及验证方法参照文献[23]。
2 结果与分析
2.1 细菌多样性、丰富度和16S rRNA基因丰度
19个站位样品高通量测序共获得697 260条序列, 平均序列长度为273 bp, 样品原始序列数在31 197~44 799之间, 抽平后序列数为27 791。97%相似水平下共获得2 375个OTU, 菲尔德斯半岛东部样品的OTU数目(Mn=954)、Shannon指数(Mn=4.83)和Chao1指数(Mn=1205)普遍高于西部样品(Mn分别为610、3.92、823)(独立样品t检验,<0.05)(表2)。每个站位的覆盖度数值(Good’s Coverage)都在98%以上, 表示本研究的采样可以较好地代表采样区域的微生物群落组成。荧光定量PCR结果显示, 西部区域的细菌丰度平均为3.49×108copies·g–1, W1站位细菌丰度最高, W4站位细菌丰度最低, 东部区域的细菌丰度平均为1.89×108copies·g–1, E11站位细菌丰度最高, 在E2、E3和E9站位细菌丰度较低。
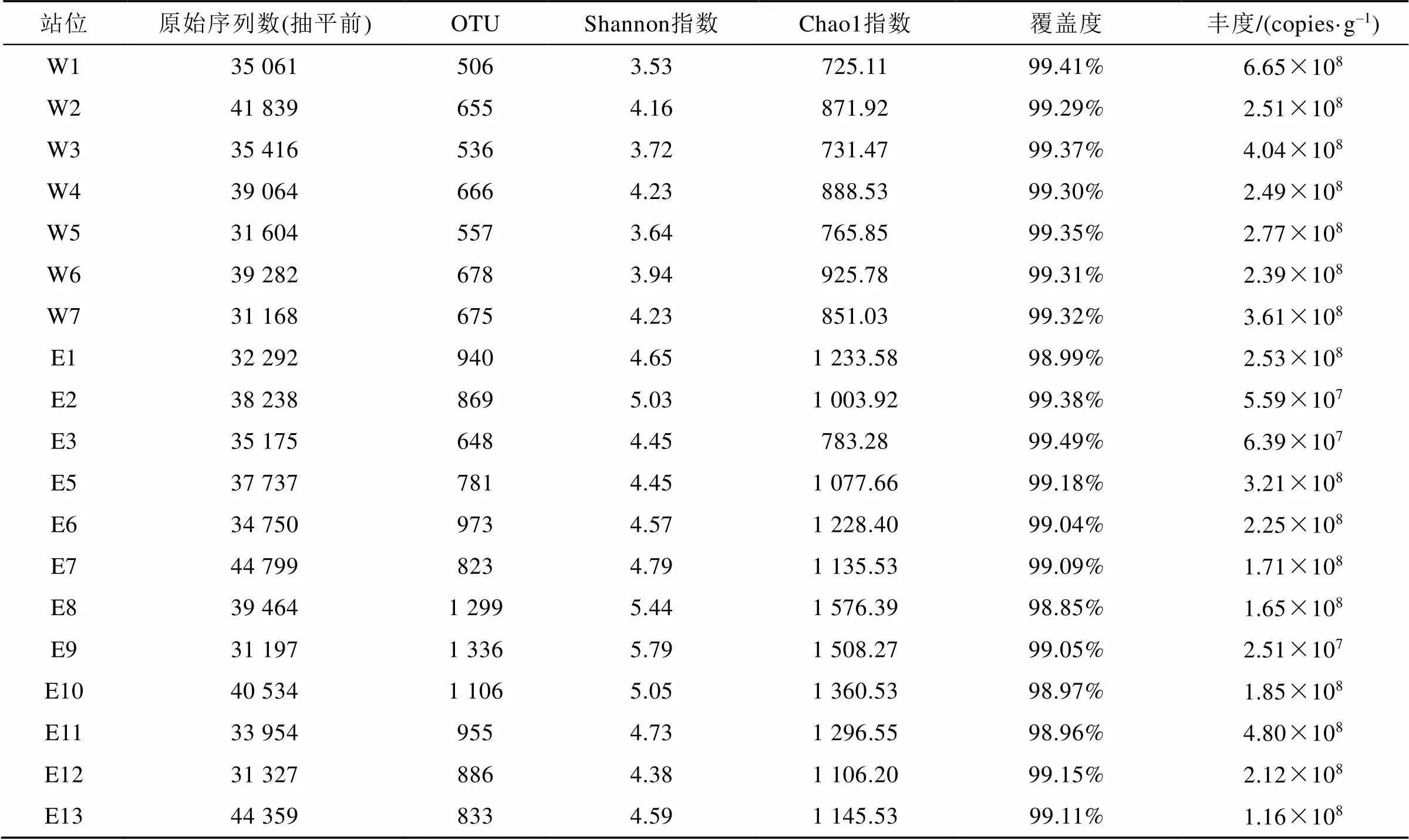
表2 基于97%相似性水平下的OTU的多样性、丰富度和16S rRNA基因绝对丰度
2.2 细菌群落组成
将19个站位的样品测序结果经过优化后得到的OTU表格基于Bray-Curtis距离进行NMDS聚类分析(图2), 可以发现沿着第一轴的方向上样品按照东西部区域进行聚类(<0.05)。将细菌群落结构和环境因子进行db-RDA分析(图3), 结果与NMDS一致, 各站位按照半岛东西部分别进行聚类。其中经度是影响样品聚类的最主要因素(=7.0,=0.002)(环境因子数据由中国海洋大学刘晓收老师提供)。
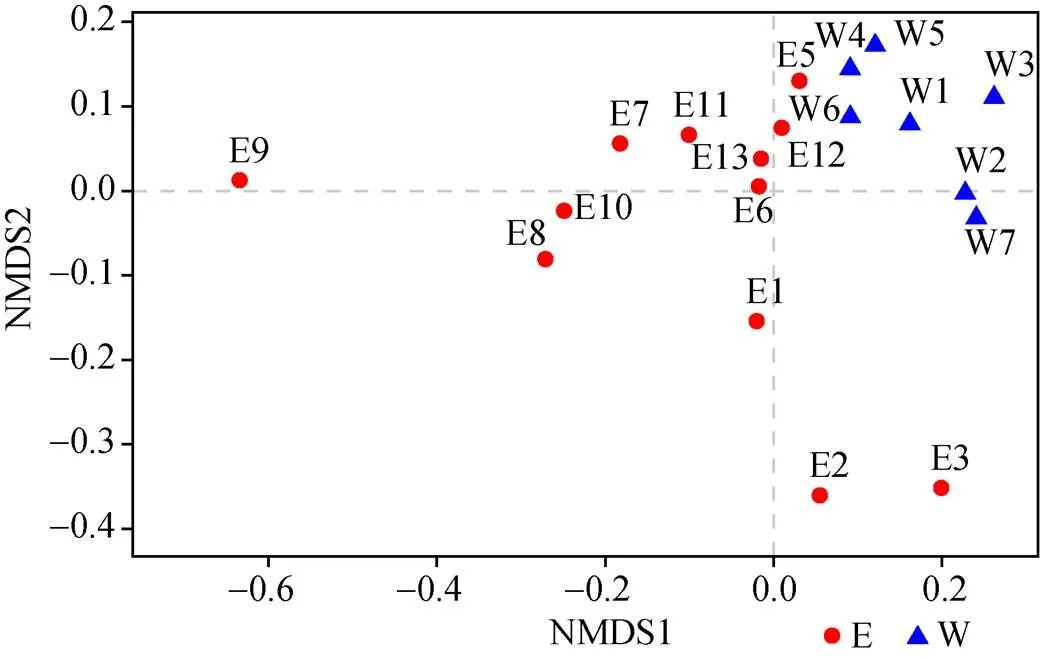
图2 OTU水平上基于Bray-Curtis距离的NMDS
Fig.2. NMDS based on Bray-Curtis distance at the OTU level
根据Silva数据库对所得的OTU序列进行分类学注释。结果表明, 本实验沉积物样品共获得42个门, 变形菌门占绝对优势, 达到40%, 其次是拟杆菌门(30.15%)、放线菌门(18.39%)、浮霉菌门(Planctomycetes, 1.83%)。这4个门的微生物占总体比例的91.54%, 是南极菲尔德斯半岛沉积物中的主要细菌类群(图4)。在变形菌门中, γ-变形菌纲占据较大比例(占总体比例的30.09%), 其次为α-变形菌纲(Alphaproteobacteria, 7.36%)。α-变形菌纲的相对丰度在东部显著高于西部(< 0.05), 且在E3站位相对丰度最高。拟杆菌门中占较高比例的为黄杆菌纲(占拟杆菌门的83.83%), 且东部区域的黄杆菌纲显著低于西部区域(< 0.05)且在E2、E3和E9站位相对丰度较低。浮霉菌门、绿弯菌门(Chloroflexi)相对丰度在半岛东部显著高于西部(<0.05), 且在E9站位具有较高的丰度。

图3 OTU水平上的db-RDA分析
Fig.3. The db-RDA analysis at the OTU level

图4 丰度占总体比例的前0.5%门水平上的群落堆积图
Fig.4. Bacterial community composition at phylum level of the more than 0.5% bacteria
在属的水平上(图5), γ-变形菌纲的颗粒状球菌属()占绝对优势(总体比例的19.97%), 其次为黄杆菌科中的一个还未被分类的类群()(8.89%)。黄杆菌科的属在东部站位普遍低于西部站位。α-变形菌纲的红细菌科(Rhodobacteraceae)与γ-变形菌纲的伍斯氏菌科(Woeseiaceae/JTB255)丰度东部站位显著高于西部站位。
2.3 可培养细菌多样性及胞外酶活性测定
2016年, 从16个站位沉积物样品共分离菌株473株, 经过酶切之后选取图谱不同的129株细菌进行测序, 测序结果显示这些细菌分属于4个门, 25个属, 50个种。丰度最高的为拟杆菌门(41株, 20个种), 其次为变形菌门(38株, 17个种)、放线菌门(31株, 8个种)和厚壁菌门(19株, 5个种)。优势属为盐细菌属()、动性球菌属(s)、莫拉氏菌属()、嗜冷杆菌属()、节杆菌属()等(图6)。优势种是阿穆斯基湾盐水杆菌()(22株)、嗜盐动性球菌()(10株)、盐晶嗜冷杆菌()(7株)等。从分离培养基来看, R2A上分离83株, 包括变形菌门27株, 15个种, 其次为拟杆菌门(25株, 12个种)、厚壁菌门(15株, 5个种)、放线菌门(16株, 6个种)。这87株菌属于21个属, 其中动性球菌属占优势, 其次为莫拉氏菌属、盐细菌属、嗜冷杆菌属等。2216E上共分离46株细菌, 其中放线菌门占优势(15株, 3个种), 其次为拟杆菌门(14株, 10个种)、变形菌门(13株, 8个种)、厚壁菌门(4株, 2个种)。46株菌分别属于17个属, 其中盐细菌属占优势, 其次为嗜冷杆菌属、动性球菌属等。R2A上分离出的细菌种类较多, 共分离44个种, 2216E上分离出24个种。在地理位置上, 东部变形菌门占优势, 其次为拟杆菌门, 在属水平上嗜冷杆菌属占优势, 其次为盐细菌属、莫拉氏菌属等。西部拟杆菌门占优势, 其次为放线菌门, 属水平上盐细菌属占优势, 其次为动性球菌属、嗜冷杆菌属等。西部细菌多样性高于东部, 呈现出地理位置上的差异。
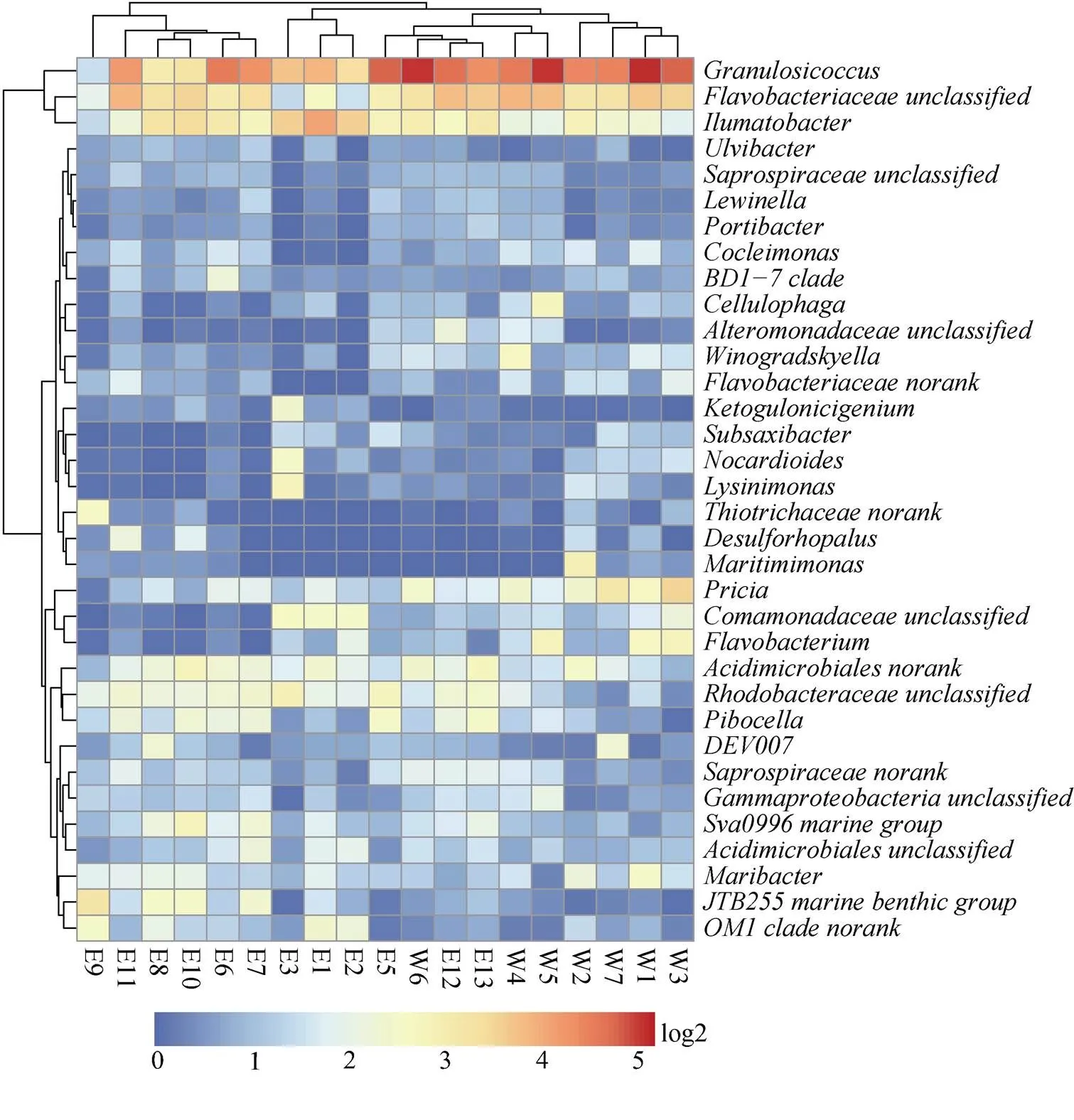
图5 丰度在总丰度前0.5%属水平的heatmap图
Fig.5. Bacterial community composion at genus level of the more than 0.5% bacteria

图6 属水平上可培养细菌多样性
Fig.6. Cultivable bacterial diversity at the genus level
依据文献报道从已测序的菌株中选取酶活研究较少的种属38株(表3)进行酶活力检测。其中24株细菌具有淀粉酶活性, 有17株可以降解吐温20, 12株可以降解吐温40, 5株可以降解吐温80, 同时可以降解吐温20和40的菌株有10株, 同时可以降解吐温20、40、80的有4株, 具有褐藻胶酶活性的有4株, 具有明胶酶活性的有18株, 具有纤维素酶活性的有4株。其中具有两种或者两种以上酶活性的菌株占55%, 表明菲尔德斯半岛地区微生物具有较广泛降解大分子物质的能力。
3 讨论
本实验所用样品为2016年中国第32次南极科考采集的南极菲尔德斯半岛潮间带19个站位的沉积物。为了研究潮间带沉积物细菌的群落组成, 我们采用16S rRNA高通量测序技术以及传统的分离培养法系统地对非可培养以及可培养细菌多样性进行研究。对高通量结果进行分析, 发现变形菌门、拟杆菌门、放线菌门为主要优势类群, 其中, 变形菌门中的γ-变形菌纲占比最高, 这与其他的研究结果基本一致[24-25]。拟杆菌门在海洋环境、土壤及沉积物中也是广泛分布的一个类群, 在本研究中也具有较高丰度(30.15%)。据报道, 该门类细菌可以降解环境中存在的大分子聚合物, 如几丁质、纤维素等[26],为其他的异养细菌提供能量和营养物质。本研究通过NMDS分析(图2)发现采取的样品基本按照东西部进行分离, 可能是因为半岛的东海岸多个国家建有科考站, 而西海岸有较多的动物栖息, 造成东西海岸微生物的分布不同[6]。在研究中我们发现位于河口附近的E2、E3以及长城站附近的E9站位微生物组成与其他站位显著不同, qPCR结果证明这3个站位细菌丰度也较低(表2)。其中黄杆菌纲在E2、E3站位丰度较低, 具体表现为黄杆菌科中的一个还未被分类的类群和属, 据报道, 海洋来源的黄杆菌具有可以编码视紫红质蛋白的基因, 使细菌在营养物质丰富或者缺乏的情况下通过光照获得能量[27]。其中在这两个站位中检测出装甲菌门(Armatimonadetes)、暗黑菌门(Atribacteria)、迷踪菌门(Elusimicrobia), 这在其他站位并没有检测到。研究表明, 暗黑菌门在缺氧的富甲烷沉积物中比较常见[28]。E9站位中绿弯菌门的厌氧绳菌科(Anaerolineaceae)和δ-变形菌纲(Deltaproteobacteria)丰度较高。厌氧绳菌科已有研究证明在烃类有机物质多的区域较为丰富[29]。δ-变形菌纲中大多数细菌属于硫酸盐氧化菌, 暗示在该站位经常发生硫酸盐氧化还原反应, 脱硫杆菌()占δ-变形菌纲的63%, 其数量会随着有机污染的增加而增多[30]。E9站位位于长城站附近, 人类活动对该站位微生物分布具有较大影响[31]。通过db-RDA分析(图3)发现样品分布规律与NMDS结果一致, 但环境因子包括温度、盐度、pH和叶绿素对各站位微生物群落聚类没有显著影响, 表明该地区影响微生物群落结构和分布的主要因素可能仍需要进一步研究。

表3 胞外酶活性检测
*菌株具有的酶活性以“透明圈直径/菌落直径”表示, -表示无活性, W表示活性微弱。
与2012年分析结果(NCBI登录号为: SRP045912)进行比较后发现, 本研究中的β-变形菌纲、疣微菌门 (Verrucomicrobia)、蓝细菌门(Cyanobacteria)、硝化螺菌门(Nitrospirae)具有相对较高的丰度, 厚壁菌门和ε-变形菌纲(Epsilonproteobacteria)具有相对较低的丰度 (<0.05), 表现出年际变化。有文献报道, 蓝细菌门的光合作用有利于沉积物颗粒形成, 促进有机碳沉淀, 在极地环境中占主导[32]。硝化螺菌门在污水处理厂具有广泛分布, 可以利用各种有机化合物及亚硝酸盐等物质[33]。这些结果表明, 相比于2012年, 菲尔德斯半岛沉积物中有机物含量可能增加, 导致相关细菌数量发生变化, 这还需要多方面的证据。
对于可培养细菌, 在先前对南极不同地区沉积物微生物多样性的研究中, 分离出的细菌分属于变形菌、拟杆菌、放线菌和厚壁菌[34-36]。本研究中分离得到的主要为变形菌, 其次为放线菌、拟杆菌, 这与以往的研究基本一致。丁新彪等[35]从普里兹湾及邻近海域沉积物中分离出较多假交替单胞菌属(), 其次为属科尔韦尔氏菌属()等。李胜男[37]从南极菲尔德斯半岛潮间带沉积物中分离出较多的假交替单胞菌属、动性球菌属、属、嗜冷杆菌属等。本研究中优势属为盐细菌属、嗜冷杆菌属、莫拉氏菌属、节杆菌属等。这与上述结果具有一定的差异, 可能与采样时间位点及培养方式有关。从分离培养基来看, R2A可以分离出较多种类的细菌, 这在2216E培养基上没有得到分离, 这也为以后分离菲尔德斯半岛可培养细菌提供思路。根据地理位置来看, 门水平上, 东部变形菌门和拟杆菌门占优势, 西部拟杆菌门和放线菌门占优势。属水平上, 东部优势类群主要为嗜冷杆菌属, 西部的优势类群主要是盐细菌属, 且西部区域的多样性高于东部区域, 可能是地理位置对细菌分布产生部分影响。
南极常年冰雪覆盖, 温度较低, 微生物能在这种寒冷的条件生存, 细菌体内关于各种代谢的低温酶起到很大的作用。在先前的研究中, 各国科学家从南极不同地区采集的样品中分离出可以在低温条件下具有较强降解酶活性的微生物菌株[38-39]。本研究在较低的温度下对38株细菌的5种酶活性进行筛选, 其中产淀粉酶的菌株占比较大, 其次为脂肪酶。低温淀粉酶在工业上主要应用于制作面食, 可以减少发酵时间, 提高口感, 还可以用于造纸、酿造及微生态制剂等多种领域。低温脂肪酶可以作为洗涤添加剂提高洗涤效果, 也可应用于造纸行业。低温条件下分离得到的微生物是生产低温酶的主要来源, 具有良好的应用潜力。
此外, 我们通过非培养方法获得的优势菌株中丰度较高的颗粒状球菌属、属、属、属并没有通过可培养的方式分离到。这可能与我们采用的有限的分离培养条件有关。这些类群大多属于寡营养类群, 且生长缓慢。有报道颗粒状球菌属、属、属可以用1/10的R2A或者2216E培养基进行分离, 而属则有专门的R培养基[40-43]。而我们所用培养基没有经过稀释, 营养物质较丰富, 因此适合这种培养条件的细菌快速生长后限制了这些寡营养细菌的生长。这提示我们在进行环境微生物分离过程中, 可以通过非培养的结果作为参考, 设计更合理的培养方案, 以提高可培养细菌的多样性, 更好地利用南极微生物资源。
1 刘春影, 丛柏林, 王能飞, 等. 南极菲尔德斯半岛可培养土壤微生物多样性及理化性质鉴定[J]. 海洋学报, 2016, 38(6): 69-81.
2 CAMPBELL I B, CLARIDGE G G C, BALKS M R. The effect of human activities on moisture content of soils and underlying permafrost from the McMurdo Sound region, Antarctica[J]. Antarctic Science, 1994, 6(3): 307-314.
3 YERGEAU E, SCHOONDERMARK-STOLK S A, BRODIE E L, et al. Environmental microarray analyses of Antarctic soil microbial communities[J]. ISME Journal, 2009, 3(3): 340-351.
4 XIAO X, LI M G, YOU Z Y, et al. Bacterial communities inside and in the vicinity of the Chinese Great Wall Station, King George Island, Antarctica[J]. Antarctic Science, 2007, 19(1): 11-16.
5 ZENG Y X, YU Y, QIAO Z Y, et al. Diversity of bacterioplankton in coastal seawaters of Fildes Peninsula, King George Island, Antarctica[J]. Archives of Microbiology, 2014, 196(2): 137-147.
6 杨晓, 丁慧, 臧家业, 等. 南极菲尔德斯半岛土壤可培养细菌多样性分析[J]. 极地研究, 2016, 28(1): 34-41.
7 林学政, 侯旭光, 李光友. 南极微生物低温酶的研究进展[J]. 海洋科学, 2002, 26(10): 38-42.
8 RAY M K, DEVI K U, KUMAR G S, et al. Extracellular protease from the Antarctic yeast Candida humicola[J]. Applied and Environmental Microbiology, 1992, 58(6): 1918-1923.
9 XIAO C, HUANG H Q, YE J J, et al.gen. nov., sp. nov., a novel member of the family Intrasporangiaceae[J]. International Journal of Systematic and Evolutionary Microbiology, 2011, 61(3): 659-664.
10 DINEEN S M, ARANDA R IV, ANDERS D L, et al. An evaluation of commercial DNA extraction kits for the isolation of bacterial spore DNA from soil[J]. Journal of Applied Microbiology, 2010, 109(6): 1886-1896.
11 PARADA A E, NEEDHAM D M, FUHRMAN J A. Every base matters: Assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples[J]. Environmental Microbiology, 2016, 18(5): 1403-1414.
12 MIAO T J, GAO S, JIANG S W, et al. A method suitable for DNA extraction from humus-rich soil[J]. Biotechnology Letters, 2014, 36(11): 2223-2228.
13 WANG X L, YAN Y G, GAO D W. The threshold of influent ammonium concentration for nitrate over-accumulation in a one-stage deammonification system with granular sludge without aeration[J]. Science of the Total Environment, 2018, 634(1): 843-852.
14 SCHLOSS P D, WESTCOTT S L, RYABIN T, et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities[J]. Applied and Environmental Microbiology, 2009, 75(23): 7537-7541.
15 CHAO A, BUNGE J. Estimating the number of species in a stochastic abundance model[J]. Biometrics, 2002, 58(3): 531-539.
16 MAGURRAN A E. Ecological Diversity and Its Measurement[M]. Dordrecht: Springer Netherlands, 1988: 1-5.
17 GOOD I J. The population frequencies of species and the estimation of population parameters[J]. Biometrika, 1953, 40(3/4): 237.
18 WANG L, LIU X S, YU S L, et al. Bacterial community structure in intertidal sediments of Fildes Peninsula, maritime Antarctica[J]. Polar Biology, 2017, 40(2): 339-349.
19 MARMUR J. A procedure for the isolation of deoxyribonucleic acid from micro-organisms[J]. Journal of Molecular Biology, 1961, 3(2): 208-218.
20 WEISBURG W G, BARNS S M, PELLETIER D A, et al. 16S ribosomal DNA amplification for phylogenetic study[J]. Journal of Bacteriology, 1991, 173(2): 697-703.
21 LANDE E S. Mapping mendelian factors underlying quantitative traits using RFLP linkage maps [J]. Genetics, 1989, 121(1): 185-199.
22 YOON S H, HA S M, KWON S, et al. Introducing EzBioCloud: A taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies[J]. International Journal of Systematic and Evolutionary Microbiology, 2017, 67(5): 1613-1617.
23 赵锐. 青岛近海两种生态环境可培养细菌多样性研究及3株海洋新菌的分类鉴定[D]. 青岛: 中国海洋大学, 2012.
24 FAN J F, LI L L, HAN J L, et al. Diversity and structure of bacterial communities in Fildes Peninsula, King George Island[J]. Polar Biology, 2013, 36(10): 1385-1399.
25 BALDI F, MARCHETTO D, PINI F, et al. Biochemical and microbial features of shallow marine sediments along the Terra Nova Bay (Ross Sea, Antarctica)[J]. Continental Shelf Research, 2010, 30(15): 1614-1625.
26 FOONG C P, LING C M W V, GONZÁLEZ M. Metagenomic analyses of the dominant bacterial community in the Fildes Peninsula, King George Island (South Shetland Islands)[J]. Polar Science, 2010, 4(2): 263-273.
27 KWON Y M, KIM S Y, JUNG K H, et al. Diversity and functional analysis of light-driven pumping rhodopsins in marine Flavobacteria[J]. MicrobiologyOpen, 2016, 5(2): 212-223.
28 CARR S A, ORCUTT B N, MANDERNACK K W, et al. Abundant Atribacteria in deep marine sediment from the Adélie Basin, Antarctica[J]. Frontiers in Microbiology, 2015, 6: 872-883.
29 TISCHER K, KLEINSTEUBER S, SCHLEINITZ K M, et al. Microbial communities along biogeochemical gradients in a hydrocarbon-contaminated aquifer[J]. Environmental Microbiology, 2013, 15(9): 2603-2615.
30 LIU J W, LIU X S, WANG M, et al. Bacterial and archaeal communities in sediments of the north Chinese marginal seas[J]. Microbial Ecology, 2015, 70(1): 105-117.
31 SANTOS I R, SILVA-FILHO E V, SCHAEFER C E G R, et al. Heavy metal contamination in coastal sediments and soils near the Brazilian Antarctic Station, King George Island[J]. Marine Pollution Bulletin, 2005, 50(2): 185-194.
32 GOLUBIC S, SEONG-JOO L, BROWNE K M. Cyanobacteria: Architects of sedimentary structures[M]//RIDING R E, AWRAMIK S M. Microbial Sediments. Berlin, Heidelberg: Springer Berlin Heidelberg, 2000: 57-67.
33 DAIMS H, LEBEDEVA E V, PJEVAC P, et al. Complete nitrification by Nitrospira bacteria[J]. Nature, 2015, 528(7583): 504-509.
34 LEE Y M, JUNG Y J, HONG S G, et al. Diversity and physiological characteristics of culturable bacteria from marine sediments of Ross Sea, Antarctica[J]. The Korean Journal of Microbiology, 2014, 50(2): 119-127.
35 丁新彪, 丛柏林, 张扬, 等. 南极普里兹湾及邻近海域沉积物微生物多样性与生理生化研究[J]. 海洋科学进展, 2014, 32(2): 209-218.
36 YU Y, LI H R, ZENG Y X, et al. Phylogenetic diversity of culturable bacteria from Antarctic sandy intertidal sediments[J]. Polar Biology, 2010, 33(6): 869-875.
37 李胜男. 南极菲尔德斯半岛潮间带沉积物细菌的多样性研究[D]. 青岛: 青岛科技大学, 2019.
38 RAY M K, DEVI K U, KUMAR G S, et al. Extracellular protease from the Antarctic yeast Candida humicola[J]. Applied and Environmental Microbiology, 1992, 58(6): 1918-1923.
39 VAZQUEZ S C, MERINO L R, CORMACK W P M, et al. Protease producing psychrotrophic bacteria isolated from Antarctica[J]. Polar Biology, 1995, 15(2): 131-135.
40 BAEK K, CHOI A, KANG I, et al.sp. nov., isolated from Antarctic seawater, and emended description of the genus[J]. International Journal of Systematic and Evolutionary Microbiology, 2014, 64(12): 4103-4108.
41 MATSUMOTO A, KASAI H, MATSUO Y, et al.gen. nov., sp. nov., a novel actinobacterium isolated from the sediment of an estuary[J]. The Journal of General and Applied Microbiology, 2009, 55(3): 201-205.
42 YU Y, LI H R, ZENG Y X, et al.gen. nov., sp. nov., a member of the family Flavobacteriaceae, isolated from Antarctic intertidal sediment[J]. International Journal of Systematic and Evolutionary Microbiology, 2012, 62(9): 2218-2223.
43 NEDASHKOVSKAYA O I, KIM S B, LEE K H, et al.gen. nov., sp. nov., a novel marine bacterium of the family Flavobacteriaceae isolated from the green alga Acrosiphonia sonderi[J]. International Journal of Systematic and Evolutionary Microbiology, 2005, 55(1): 177-181.
BACTERIAL COMMUNITY STRUCTURE OF INTERTIDAL SEDIMENTS IN THE FILDES PENINSULA, ANTARCTICA, AND PRELIMINARY SCREENING OF ENZYME-PRODUCING STRAINS
Wu Leilei1, Shang Li1, Sun Hao1, Shi Xiaochong1,2, Zhang Xiaohua1,2
(1College of Marine Life Sciences, Ocean University of China, Qingdao 266003, China;2Laboratory of Marine Ecology and Environmental Science, Pilot National Laboratory for Marine Science and Technology, Qingdao 266071, China)
To study the composition of the bacterial community and enzyme-producing activity of cultivable bacteria in the intertidal sediments of the Fildes Peninsula, Antarctica, sediment samples from 19 sites of the intertidal zone were collected, and the bacterial community composition of these sediments was sequenced by high-throughput sequencing using Illumina MiSeq technology. The absolute abundance of total bacteria in the sediments was measured by real-time PCR. The plate coating method was used to purify and isolate the culturable bacteria of sediment samples from 16 sites. Thirty-eight strains of bacteria were selected for the screening of enzyme activities. In total, 2 375 operational taxonomic units (OTUs) of the intertidal sediment samples were obtained, and these OTUs were assigned into 42 bacterial phyla, of which Proteobacteria, Bacteroidetes, and Actinobacteria were the dominant groups. The abundance of total bacteria ranged from 2.51 × 107to 6.65 × 108copies·g−1. Among culturable bacteria, 129 strains were assigned to four phyla, 25 genera, and 50 species. Proteobacteria, Actinobacteriaand Bacteroides were the predominant groups, which were basically consistent with the results of high-throughput sequencing. Thirty-eight strains were selected for the screening of low-temperature enzyme activities (including lipase, amylase, gelatinase, alginate, and cellulase), and 17 strains were found to produce lipase, 24 strains produced amylase, 18 strains produced gelatin, four strains produced alginate, and four strains produced cellulase.
intertidal sediments of the Fildes Peninsula, high-throughput sequencing, culturable bacteria, 16S rRNA gene, enzyme activity
2019年11月收到来稿, 2019年12月收到修改稿
国家海洋局极地专项(CHINARE-04-01、CHINARE-02-01)资助
吴蕾蕾, 女, 1996年生。硕士生, 主要研究方向为南极微生物多样性。E-mail: wuleilei0524@163.com
史晓翀, E-mail:shixiaochong@ouc.edu.cn
10. 13679/j.jdyj.20190069
猜你喜欢
今日农业(2021年8期)2021-11-28
水上消防(2021年3期)2021-08-21
中国比较医学杂志(2020年4期)2020-05-26
中国报道(2020年12期)2020-01-08
人大建设(2019年5期)2019-10-08
水生生物学报(2019年4期)2019-07-20
人大建设(2019年3期)2019-07-13
生物安全学报(2019年3期)2019-02-15
川北医学院学报(2019年6期)2019-02-10
中国自行车(2018年7期)2018-08-14
