
重组猪胰蛋白酶原及其突变体在毕赤酵母X33中的组成型表达
2021-01-05 08:40:14窦俊方宏清徐登圆赵珊珊陈慧梅徐晓峰支艳艳李晓稳文良柱
生物技术通讯 2020年5期
窦俊,方宏清,徐登圆,赵珊珊,陈慧梅,徐晓峰,支艳艳,李晓稳,文良柱
1.中国药科大学,江苏 南京 211198;2.江苏万邦医药科技有限公司,江苏 徐州 221004;3.江苏万新医药科技(苏州)有限公司,江苏 苏州 215000
巴斯德毕赤酵母(Komagataella phaffii,曾称Pichia pastoris)是一种常用的甲基营养型酵母,近年来成为备受关注的外源蛋白真核表达系统[1],成功表达了超出5000个外源蛋白[2]。美国FDA于2006年批准毕赤酵母是安全的微生物(generally recognized as safe,GRAS)[3],其被广泛用于表达酶、膜蛋白、抗原、抗体和调节蛋白等多种外源蛋白[4-5]。相比原核表达系统,酵母表达系统具有筛选方法成熟[6]、生长快、外源蛋白表达量高、稳定性好[7]、可翻译后加工修饰[8]、可基因改造[9]、产物可分泌、可高密度发酵[10]等优点。
启动子是外源蛋白表达系统中影响外源蛋白高效表达的重要基因表达调控的顺式元件。其中,醇氧化酶 1(alcohol oxidase 1,AOX1)基因的启动子PAOX1是毕赤酵母表达系统最常用的甲醇诱导型表达的强启动子[11],广泛用于酵母的高密度、大规模发酵培养,表达了众多外源蛋白。
但是,严格受甲醇诱导表达的启动子PAOX1仍有一些不足之处。甲醇易挥发,大规模补料控制工艺复杂[12],操作过程不便;高浓度的甲醇易燃,可能有火灾的安全隐患[13];甲醇有毒,不宜于饲料工业、食品、医药等蛋白生产,容易污染环境。
目前,甘油醛三磷酸脱氢酶(glyceraldehydes-3-phosphatedehydrogenase,GAP)启动子和 pYPT1(GTP酶)启动子是应用较为广泛的组成型启动子。PGAP可利用的碳源有葡萄糖、甘油、甲醇和油酸等,克服了PAOX1启动子的缺陷,高水平组成表达相当甚至高于PAOX1表达系统的外源蛋白[14]。比起PAOX1的诱导型表达,使用PGAP的组成型表达不需要补加甲醇进行诱导,避免了甲醇带来的危害,且操作方便,大大缩短了发酵周期,提高了生产效率[15],更适合大规模连续表达外源蛋白[16],应用前景广阔。
黄鹏等[8]利用PGAP在毕赤酵母中表达了高纯度和高活性的重组人鹅型溶菌酶2(rhLysG2);Yang等通过PGAP表达了碱性植酸酶[17];方炜等[18]构建菌株 GS115(pGAP9K-AMPD),表达了 AMP脱氨酶;王金菊等[19]构建毕赤酵母GS115(pGAP9-16BMP)进行组成型表达,得到了目的产物猪肉风味强化肽。
胰蛋白酶原可被肠激酶或胰蛋白酶自身激活成为活化的胰蛋白酶[20]。胰蛋白酶的相对分子质量为 2.3×104~2.4×104,能专一性水解多肽链中赖氨酸和精氨酸C端形成的肽键。胰蛋白酶在皮革处理、食品加工、医药、临床诊断、蛋白质组学和重组胰岛素的生产等领域应用广泛[21]。传统工艺从动物(猪、猪等)胰脏中提取的胰蛋白酶纯度低,可能携带病原体或病毒,易发生自溶[22],价格昂贵,在制药行业中应用受限[23]。在大肠杆菌和毕赤酵母中表达的重组胰蛋白酶原[24],无外源性病毒污染,无杂酶,酶切反应的特异性较高,无其它副反应[25],解决了上述问题。徐晓峰等通过构建含胰蛋白酶原基因的质粒,转化大肠杆菌BL21(DE3),以包涵体形式表达,经过变性复性和激活后得到了胰蛋白酶[26];汪晓娟等构建了启动子为PAOX1的质粒,电转化毕赤酵母GS115,诱导表达获得了猪胰蛋白酶[20]。
本实验中,我们采用基因重组技术,利用PGAP组成型启动子,无需添加甲醇诱导,实现了猪胰蛋白酶原基因在毕赤酵母X33中的重组分泌表达,获得了活性较高的重组猪胰蛋白酶,简化了操作,缩短了表达周期,提升了效率,避免了动物源性污染,为后续实验奠定了基础。
1 材料与方法
1.1 材料
大肠杆菌DH5α和毕赤酵母X33均为本实验室保藏;质粒pGAP-PT、pGAP-PT1、pGAP1-PT1、pGAP2-PT1、pGAP3-PT1、pGAP4-PT1为组成型表达胰蛋白酶原或其突变体的酵母表达载体,委托南京金斯瑞生物科技有限公司构建,由本实验室保存。
限制性内切酶AvrⅡ(New England BioLabs);CaCl2、山梨醇、N-苯甲酰-L-精氨酸乙酯盐酸盐、磷酸、乙腈(国药集团);博来霉素(zeocin,Invitrogen);琼脂糖(Generay Biotech);LB抗性培养基含25 μg/mL博来霉素;YPD抗性培养基含 100 μg/mL博来霉素。
组合式摇床(Qiang Le);高速落地冷冻离心机(Thermo Scientific);紫外分光光度计UV-2450、基因导入仪SCIENTZ-2C(宁波新芝);生物超净工作台(SUKUN);立式压力蒸汽灭菌器(上海博迅);ÄKTAexplorer(GE);隔水式恒温培养箱(上海一恒);吸光度酶标仪Apollo 11 LB913(Berthold Technologies Bioanalytic);LC-MS(安捷伦);苯甲醚亲和层析柱、SP离子交换柱(中科森辉);HPLC仪(岛津)。
1.2 质粒扩增
将质粒采用冷CaCl2法转化大肠杆菌DH5α,涂布于含25 μg/ml博来霉素的LB固体培养基平板上,37℃倒置培养至菌落形成。筛选阳性克隆点到LB液体培养基中,以37℃、250 r/min的条件培养约16 h,用40%甘油于-20℃保存菌种,并转接20 μL菌液至200 mL LB液体培养基中,37℃、250 r/min培养约16 h。
1.3 质粒提取、线性化与回收
采用NucleoBond Xtra Midi kit for transfection-grade plasmid DNA-REF 740410.50质粒中抽试剂盒提取上述菌液的质粒,测定质粒含量,并取 20 μg质粒,加入 6 μLAvrⅡ酶,建立总体积为400 μL的线性化酶切体系,37℃孵育2 h。
取少量样品进行DNA琼脂糖电泳验证,结果符合要求后用TaKaRa MiniBEST DNA Fragment Purification Kit Ver.4.0-Cat.9761试剂盒回收,最后一步用ddH2O洗脱。
1.4 毕赤酵母X33感受态制备
具体方法参见文献[27-28]。
1.5 毕赤酵母X33的电击转化和筛选
用基因导入仪将构建的载体与毕赤酵母X33感受态细胞混合,转入电转杯,冰浴5 min,将电转化杯置于电转仪中,启动RD键开始电击。转化结束后立即加入1 mol/L冰冷的山梨醇1 mL,涂布于含100 μg/mL博来霉素的YPD平板进行筛选,30℃培养2~4 d至长出单菌落。
1.6 SDS-PAGE分析
筛选单菌落到3 mL YPD培养基中,30℃、280 r/min培养20~22 h,按起始D600nm为0.05转接到50 mL YPD培养基中,30℃、250 r/min培养48 h,取1 mL培养液,12 000 r/min离心3 min,取上清加非还原处理液进行SDS-PAGE,观察结果。
1.7 活性测定
首先采用紫外-可见分光光度法测定浓度。以 0.01 mol/L HCl、0.02 mol/L CaCl2(pH2.0±0.2)缓冲液为空白对照,将样品用缓冲液溶解或稀释至约0.5 mg/mL,取光程为1 cm的带盖石英比色杯,测定D280nm值。按下式计算蛋白浓度:
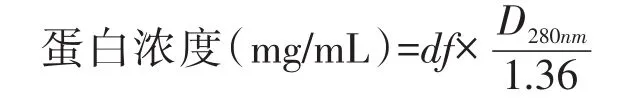
式中,df为供试品稀释倍数,D280nm为供试品溶液在280 nm下扣除空白对照后的吸收值。
然后采用BAEE法,参照中国药典2015版2部[29]测定酶活力。含有0.25 mmol/L BAEE底物的磷酸盐缓冲液,光程1 cm,波长253 nm,温度25.0±0.5℃,测量体积3.2 mL条件下吸光值每分钟改变0.003相当于1个酶活力单位。
1.8 胰蛋白酶的纯化
活化后的菌种按起始D600nm为0.2转接到含1 L YPD的5 L锥形瓶中,30℃、220 r/min培养48 h,5000 r/min离心15 min收集上清液,设置UV检测波长280 nm,用SP-FF离子交换介质捕获胰蛋白酶原,然后加入肠激酶进行激活得到胰蛋白酶,再通过苯甲醚亲和层析得到纯度较高的胰蛋白酶。
1.9 HPLC分析
1.9.1 色谱条件与系统适用性试验 用十八烷基硅烷键合多孔硅胶填充色谱柱(ODS柱),柱直径4.6 mm,长25 cm;粒度 3 μm,孔径200 Å,柱温40℃。流动相A液为水+0.1%磷酸,B液为乙腈+0.1%磷酸,流速1.0 mL/min,检测波长280 nm。洗脱梯度为 0→25→30→34min→35→45 min,对应时间点的流动相B为25%→45%→90%→90%→25%→25%。α胰蛋白酶和β胰蛋白酶的分离度应不小于1。
1.9.2 供试品溶液制备 取适量重组胰蛋白酶,用 0.01 mol/L HCl、0.02 mol/L CaCl2(pH2.0±0.2)溶液配制成70±10 mg/mL,转移至HPLC进样瓶。
1.9.3 测定法 取供试品溶液1 μL注入液相色谱仪,Chromeleon 7.2记录色谱图。面积归一化法计算胰蛋白酶纯度,积分时间为25 min,扣除空白对照。α胰蛋白酶如有前肩峰采用垂直积分,β胰蛋白酶如有拖尾峰采用切线积分,β胰蛋白酶不低于70%,α胰蛋白酶不高于20%。
1.10 胰蛋白酶LC-MS分析
采用液质联用仪进行质谱分析。流动相A为超纯水+0.1%甲酸,B为ACN+0.1%甲酸。色谱柱为 Sepax Bio-C18,4.6 mm×250 mm,5 μm,300 Å。流速1.0 mL/min,柱温35℃,检测波长214 nm。洗脱梯度为0→35→37→38→40 min,对应时间点的流动相B为10%→90%→90%→10%→10%。
2 结果
2.1 猪胰蛋白酶原及其突变体的组成型表达菌株筛选
筛选到的8株X33/pGAP-PT菌株表达上清的SDS-PAGE显示其中7株在相对分子质量25×103附近有目的蛋白表达,菌株X33/pGAP-PT2和X33/pGAP-PT3表达比较明显(图1),考马斯亮蓝法测得X33/pGAP-PT3表达上清浓度为0.088 mg/mL,肠激酶激活后的酶活为39 U/mL。
筛选到的突变体菌株X33/pGAP-PT1表达上清在相对分子质量25×103附近有明显的目的蛋白(图2)。考马斯亮蓝法测得X33/pGAP-PT1-1表达上清浓度为0.115 mg/mL,肠激酶激活后的酶活为130 U/mL,提高了胰蛋白酶原的表达量(图3)和酶活性。
2.2 不同启动子对表达的影响
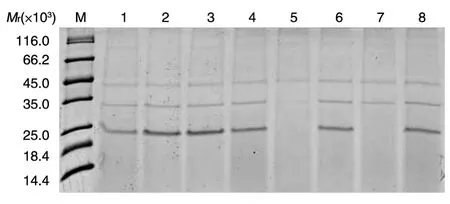
图1 X33/pGAP-PT表达上清的SDS-PAGE

图2 X33/pGAP-PT1表达上清的SDS-PAGE
突变体在胰岛素原酶切实验中胰岛素得率较高,因此,我们研究了使用 PGAP、PGAP1、PGAP2、PGAP3和PGAP4启动子的突变体的胰蛋白酶原表达情况。不同启动子突变体的表达上清SDS-PAGE显示有目的蛋白表达,且X33/pGAP1-PT1、X33/pGAP4-PT1表达明显(图4)。X33/pGAP1-PT1表达上清蛋白含量最高,考马斯亮蓝法测得浓度为0.123 mg/mL,证明更换的PGAP1启动子提高了胰蛋白酶原的表达量。
2.3 活性测定
分别将 X33/pGAP-PT、X33/pGAP-PT1、X33/pGAP1-PT1、X33/pGAP2-PT1、X33/pGAP3-PT1 和X33/pGAP4-PT1甘油菌种活化后按D600nm为0.05转接到50 mL YPD中,30℃、250 r/min培养48 h,离心取上清,加肠激酶激活,BAEE法测上清酶活性,用UV-2450及配套软件采集数据(图5)。X33/pGAP-PT、X33/pGAP-PT1、X33/pGAP1-PT1、X33/pGAP2-PT1、X33/pGAP3-PT1、X33/pGAP4-PT1表达的胰蛋白酶活性依次为39、130、142、72、62、91 U/mL,X33/pGAP1-PT1最高,证明更换的PGAP1启动子提高了胰蛋白酶活性。
2.4 胰蛋白酶的纯化
将X33/pGAP1-PT1进行5 L发酵,离心取上清,对表达上清的胰蛋白酶原进行捕获、激活和纯化,从色谱图(图6)可以看到,通过苯甲醚亲和层析得到了纯度较高的胰蛋白酶,测得浓度为0.94 mg/mL,比活6365 U/mg,活性5983 U/mL。

图3 X33/pGAP-PT与X33/pGAP-PT1上清SDS-PAGE对比

图4 X33不同启动子表达上清的SDS-PAGE
2.5 HPLC分析
对X33/pGAP1-PT1纯化后的样品进行HPLC分析,主峰保留时间15.387 min,首先被洗脱出来的是α胰蛋白酶,且不高于20%,然后是β胰蛋白酶,且不低于70%,分离度不小于1(图7)。
2.6 胰蛋白酶的LC-MS分析

图5 BAEE法酶活曲线
对X33/pGAP1-PT1纯化后的样品进行LC-MS分析,质谱测定的相对分子质量为23464.08,根据氨基酸序列推测的相对分子质量为23475.62。考虑到分子中有6对二硫键,相对分子质量减少12,所以理论值为23463.62,测定值与理论值基本一致(图8)。

图6 X33/pGAP1-PT1纯化图
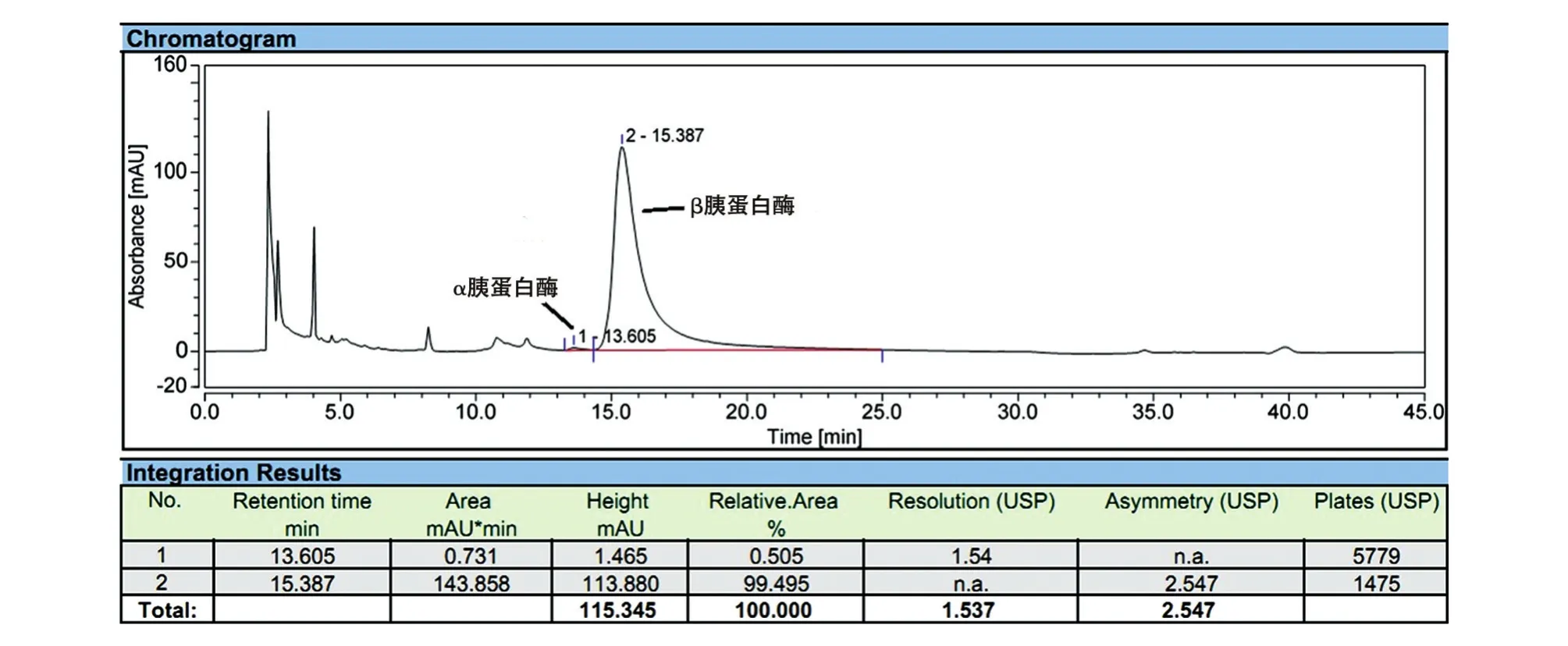
图7 胰蛋白酶的HPLC图
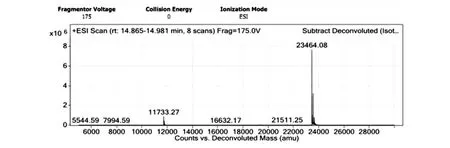
图8 X33/pGAP1-PT1的LC-MS图
3 讨论
毕赤酵母表达系统由于自身优势被广泛用于表达各种外源蛋白,本研究采用PGAP组成型启动子,避免了甲醇的使用,安全方便。胰蛋白酶是常用的消化酶,在重组胰岛素生产等领域起重要作用。本研究实现了猪胰蛋白酶原在巴斯德毕赤酵母X33中的高效组成型表达,而且产物活性较高,为后续实验奠定了基础。
利用毕赤酵母表达系统大规模发酵,经过简单几步纯化即可获得高纯度的胰蛋白酶,同时有效避免了动物源性污染。在蛋白质组学中,通常需要序列级的胰蛋白酶来特异性切割蛋白质为多肽,因此选择合适的胰蛋白酶非常重要。胰蛋白酶还可用作药物,其注射剂能消化溶解变性蛋白,但对正常组织不起作用,临床应用中能使脓液、瘤液、血凝块被分解,使引流通畅,创面加速净化,新生肉芽组织生长,还可分解破坏多种毒蛇毒液中的蛋白质用于解蛇毒。综上所述,本研究有较好的经济效益和应用前景。
猜你喜欢
舰船科学技术(2022年11期)2022-07-15 07:51:56
西藏农业科技(2019年3期)2019-11-04 00:35:10
食品与发酵工业(2018年3期)2018-04-12 09:35:51
现代园艺(2018年3期)2018-02-10 05:18:12
上海农业学报(2017年3期)2017-04-10 12:39:12
安徽医科大学学报(2016年12期)2017-01-15 14:21:44
山东农业工程学院学报(2016年6期)2016-12-01 05:38:19
广东饲料(2016年1期)2016-12-01 03:43:01
工业微生物(2016年5期)2016-11-11 06:58:44
化学与生物工程(2016年10期)2016-11-10 06:01:35
