
全二维气相色谱-电子捕获检测器测定复杂基质土壤中24种有机氯和拟除虫菊酯类农药
2020-12-28 08:19时磊孙艳艳沈小明吕爱娟蔡小虎刘娇沈加林
岩矿测试 2020年6期
时磊,孙艳艳,沈小明,吕爱娟,蔡小虎,刘娇,沈加林
(中国地质调查局南京地质调查中心,江苏 南京 210016)
有机氯农药(OCPs)是一种持久性有机污染物(POPs),由于其强烈的“三致”作用和持久稳定性,对人类健康和环境的危害极大[1-5]。在20世纪70年代,西方发达国家已经开始禁用 OCPs,我国也于1983年禁止其生产和使用[6-8],但由于使用量大,在环境中降解缓慢、滞留时间长,使得OCPs仍然是在环境中检出率最高的一类POPs[9]。拟除虫菊酯类农药是我国代替有机氯农药和其他剧毒长残留杀虫剂的主要农药类型之一,其品种数和使用量仅次于有机磷农药,占杀虫剂市场的第二位[10-16]。它的开发被称作杀虫剂农药的一个新的突破,被认为是杀虫剂历史上的第三个里程碑[17]。但菊酯类农药与OCPs存在相似的神经系统作用机制,都有一定蓄积性,以及致癌、致畸、致突变作用[18-23]。
全二维气相色谱是20世纪90年代发展起来的一种分析方法,是利用两根色谱柱间的极性差异,实现待测物质的正交分离,能够较好地解决复杂样品的分离和定性问题[24-26]。该方法凭借其高峰容量、高分辨率、高灵敏度等特点,近几年被广泛应用于各类复杂样品的分析。张兵等[27]建立了全二维气相色谱-电子捕获检测器(GC×GC-μECD)检测土壤中23种毒杀芬同类物的方法,检测灵敏度明显高于全二维气相色谱飞行时间质谱(GC×GC-TOFMS)。时秋娜等[28]应用全二维气相色谱结合质谱对动植物油中的37种脂肪酸甲酯进行了测定,该法与普通气相色谱-质谱法相比,分离效果更好。Silva等[29]应用GC×GC-TOFMS在巴西凝析油中不仅鉴定出单金刚烷和双金刚烷,还对三金刚烷和四金刚烷类化合物作了定性和定量研究。由于μECD对OCPs和拟除虫菊酯具有很高的灵敏度[30-31],将其与全二维色谱结合使用,既可以保证较高的灵敏度,又能解决单一色谱柱峰容量有限的问题,从而适用于复杂基质样品的检测。
本方法在EPA8081a和《土壤和沉积物 有机氯农药的测定 气相色谱-质谱法》(HJ 835—2017)基础上,以污染区农田土壤为研究对象,将含干扰物多且难以净化去除的土壤定义为“复杂基质土壤”,优化了全二维分析参数,通过双色谱柱串联扩大了峰容量,有效分离了干扰物,建立了应用GC×GC-μECD分析20种OCPs和4种拟除虫菊酯类农药的测定方法。
1 实验部分
1.1 仪器和主要试剂
Agilent 6890N型气相色谱仪,配有电子捕获检测器(μECD);全二维气相色谱固态热调制器(Canvas SSM1800);索氏抽提器250mL;HH-1-6型恒温水浴锅;KL512J 数控氮吹浓缩仪;DLSB-10/20型低温冷却液循环泵;WH-3型漩涡振荡仪。
有机氯农药20种混标(Supelco,47426-U):α-六六六、γ-六六六、β-六六六、七氯、δ-六六六、艾氏剂、环氧七氯、γ-氯丹、α-氯丹、硫丹Ⅰ、滴滴伊、狄氏剂、异狄氏剂、滴滴滴、硫丹Ⅱ、p,p’-滴滴涕、异狄氏剂醛、硫酸硫丹、甲氧滴滴涕、异狄氏剂酮,质量浓度2000μg/mL;胺菊酯、联苯菊酯、甲氰菊酯、氟氯氰菊酯,质量浓度100μg/mL;替代物2,4,5,6-四氯间二甲苯(Supelco),质量浓度为500μg/L;无水硫酸钠(分析纯,400℃烘烤4h);正己烷、丙酮均为色谱纯;铜片(经稀硝酸活化);弗罗里硅土净化柱(1000mg/6mL);高纯氮气(>99.999%)。
1.2 仪器工作条件
1.2.1全二维气相色谱条件
一维色谱柱(TG-35MS,30m×0.25mm×0.25μm),二维色谱柱(DB-1,0.8m×0.18mm×0.18μm)。进样口温度250℃,ECD检测器温度315℃。载气为氮气(纯度99.999%),载气流量1.0mL/min,不分流进样,进样量1.0μL。升温程序:初始温度150℃,以3℃/min升温至240℃,以4℃/min升温至300℃,后运行温度310℃,保持3min。以保留时间定性和峰面积外标法定量。
1.2.2GC-MS验证实验条件
气相色谱条件:TG-5SILMS石英毛细管柱,进样口温度 280℃,高纯氦气载气(>99.999%),恒流方式,流速1.2mL/min,不分流进样,进样量 1μL。升温程序:初始温度120℃,保持2min,以12℃/min升温至180℃,保持5min,以5℃/min升温至280℃,保持5min。质谱条件:电子轰击电离(EI)模式,电离能量70eV,离子源温度280℃,传输线温度300℃,全扫描Scan或选择离子模式(SIM)。
1.3 实验方法
1.3.1样品制备
土壤样品为2018年全国农用地详查样品,主要采自江苏扬州、宿迁等地,样品运送至实验室后放置于零下10℃冰柜保存,室温下解冻后置于搪瓷盘中,除去石子、枝片等异物,充分混合均匀。称取10.0g样品,加入适量无水硫酸钠,掺拌均匀,研磨成细粒状。同时另取土壤样品一份,用于含水率测定。
1.3.2提取与净化
将研磨均匀的样品全部装入滤纸套筒中,放入索氏提取器中,加入正己烷-丙酮(1∶1,V/V)混合溶剂200mL,提取10h,回流速率控制在6次/h左右,冷却后收集所有提取液待净化。
将活化后的铜片加入提取液放置4~6h以除去硫化物,如铜片全部变黑需补加,直至铜片不变色为止。将玻璃漏斗上垫一层玻璃棉,加入约 5g 无水硫酸钠,将提取液过滤至浓缩器皿中。再用少量正己烷-丙酮混合溶剂洗涤提取容器3次,洗涤液并入漏斗中过滤,最后再用少量正己烷-丙酮混合溶剂冲洗漏斗,全部收集至浓缩器皿中,氮吹浓缩至约0.5mL后经弗罗里硅土固相萃取小柱净化,净化柱上端加入1~2g无水硫酸钠,净化前需对固相萃取柱进行活化:依次使用正己烷-丙酮(9∶1,V/V)和正己烷各10mL对固相萃取柱进行洗脱。活化完毕后上样,用正己烷-丙酮(9∶1,V/V)混合液15mL进行洗脱、40℃下氮吹浓缩、正己烷定容至1mL,按仪器工作条件进行测定。
1.3.3质量控制
每批样品(不超过 20 个样品)需做1个空白样、1个平行样、1个空白加标和1个基质加标样。空白样品的测定结果中目标物浓度不应超过方法检出限;平行样测定结果相对偏差应小于35%;空白加标和基质加标样品回收率控制范围为60%~140%;替代物为2,4,5,6-四氯间二甲苯,回收率控制范围为60%~140%;进样口惰性检查:滴滴涕到滴滴伊和滴滴滴的降解率应不超过15%。如果滴滴涕衰减过多或出现较差的色谱峰,则需要清洗或更换进样口,同时还要截取毛细管前端的5cm,重新校准。
2 结果与讨论
2.1 提取方法的优化
选择苏北地区农田土壤作为实验对象,由于含水量较大,提取前需加入大量无水硫酸钠混合均匀,采用超声或振荡方式提取,基体容易结块,严重影响提取效率。实验比较了索氏提取、加速溶剂提取和超声波提取3种方式的提取效率。选择空白样品分成两组,每组3份进行回收实验,一组添加浓度为1.0μg/kg,另一组添加浓度为20.0μg/kg。以正己烷-丙酮(1∶1,V/V)为提取溶剂,按上述3种方法进行提取,结果表明:索氏提取不论是高浓度还是低浓度都表现出较高的回收率,分别为80.7%~94.4%和82.7%~103.5%(表1)。加速溶剂萃取效率总体与索氏提取相当,但由于个别高浓度样品含水率大,带来的管路残留难以消除,需要多次清洗才能满足后续实验。所以,最终选择索氏提取为实验的提取方法,通过实验比较索氏提取时间和提取效率的关系,按每小时回流6次左右的提取速率,提取10h,效率基本达到饱和。另外,索氏提取主要为过夜提取,节省了大量工作时间,尤其是承担大批量样品时,能大大提高工作效率。
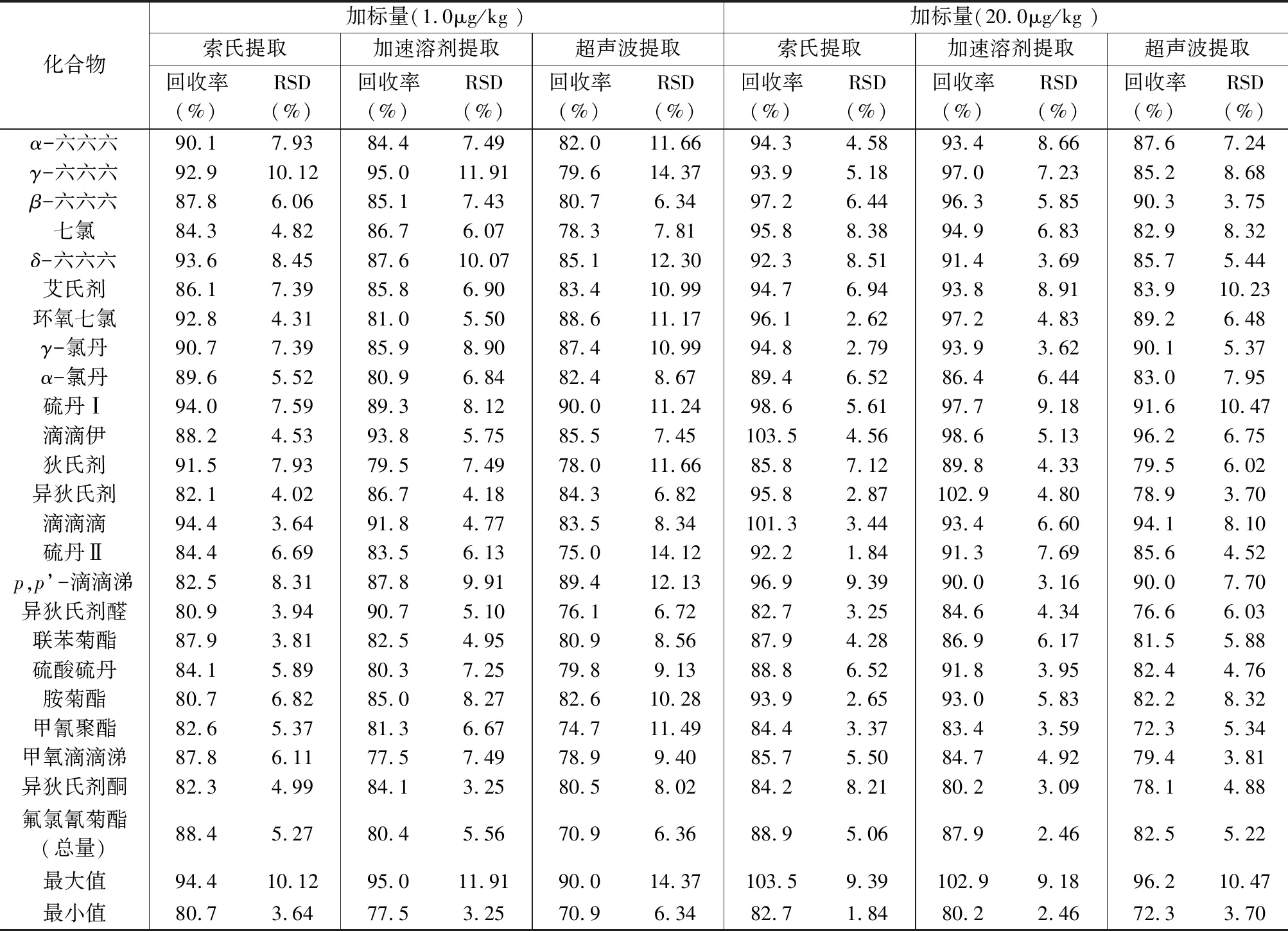
表1 方法精密度和回收率实验Table 1 Precision and recovery tests of the method
2.2 净化条件的优化
工业区农业土壤中不仅有机质含量多,而且常含有硫化物,硫化物的存在不仅会污染进样口和色谱柱,还会使ECD过载,将目标物的色谱峰掩盖,严重影响定性定量分析。实验时先将活化后的铜片加入提取液静置3~4h以除去硫化物(如铜片变黑,需补加至不变色为止);浓缩后用商品弗罗里硅土固相萃取小柱(填料6cm)净化,由于部分样品含水率较高,浓缩后明显含有水分,需在净化柱上端补加1~2g无水硫酸钠除去剩余的水分,最后用正己烷-丙酮(9∶1,V/V)混合液15mL进行洗脱。
2.3 调制周期的选择
全二维气相色谱的调制器主要起浓缩、聚焦、再进样的作用,合适的调制周期有利于化合物更好地分离,降低目标物的检出限。如果调制周期过短,会使第一维流出的组分未能在同一周期内完成调制就进入下一周期,影响目标物的定性和定量;如果调制周期过长,减少了一维峰的切割次数,易出现共流出现象,从而导致一维色谱分辨率下降。所以在满足分离度的条件下,应选择尽可能短的调制周期。本实验考察了调制周期分别为1s、2s、3s、4s时的分离效果,发现当调制周期为4s时,全部目标物均能在同一周期流出且分离度最好,故选择调制周期为4s(图1d)。
2.4 全二维气相色谱分离柱的选择
由于目标化合物较多,土壤样品基质复杂,各物质完全分离的难度很大,因此选择极性柱与非极性柱搭配的方式。待测物在极性的一维柱上,按沸点规律分离,而流出物经调制器聚焦后迅速升温送入二维柱,并在二维色谱柱上按物质极性的差异分离。实验选择3根不同极性的一维色谱柱(TG-35MS,30m×0.25mm×0.25μm;HP-1701,30m×0.25mm×0.25μm;HP-5,30m×0.25mm×0.25μm)分别与非极性二维色谱柱(DB-1,0.8m×0.18mm×0.18μm)进行组合。结果表明,TG-35MS柱和DB-1柱组合分离效果最好,每个待测目标物都有独立的一维和二维的保留时间,另两种组合分离效果相对较差,容易出现多个峰的重叠。因此,确定采用TG-35MS柱和DB-1柱组合的方式建立分析方法。
2.5 分析技术评价
2.5.1标准曲线和检出限
按仪器工作条件测定20种有机氯农药和4种拟除虫菊酯混合标准溶液,浓度为50.0μg/L,采用外标法定量,在优化的条件下获得的全二维气相色谱图分离效果好(图1d)。按色谱条件对1.0、5.0、10.0、20.0、50.0、100.0、200.0、500μg/L的混合标准溶液进行测定,以各化合物的质量浓度为横坐标,对应的峰面积为纵坐标绘制标准曲线,其中氟氯氰菊酯存在多个异构体,本实验以其质量浓度对应的多个异构体的总峰面积来计算总量。结果表明:各物质的质量浓度均在1.0~500.0μg/L范围内呈线性,相关系数(r)为0.995~0.998。在10.0g石英砂样品中加入低浓度20种有机氯农药和4种拟除虫菊酯混合标准溶液,平行7份,按实验方法进行测定,计算标准偏差s,按照检出限为3.14s计算,各物质检出限为 0.02~0.17μg/kg(结果见表2),远低于HJ 835—2017标准中各物质的检出限20.0~90.0μg/kg。马玲等[32]采用超声波提取-硫酸净化-气相色谱/质谱法同时测定土壤样品中23种有机氯农药,方法检出限为0.10~4.0μg/kg;李俊等[13]使用加速溶剂萃取-气相色谱/质谱法(选择离子监测模式)同时测定土壤中拟除虫菊酯类农药,检出限为0.11~3.4μg/kg。本方法检出限较低的原因主要是由于索氏提取效率高并且稳定性好,另一方面全二维气相色谱与电子捕获检测器结合具有良好的灵敏度。Senar等[33]使用超声波提取结合气相色谱电子捕获检测器测定土壤中的有机氯类农药,检出限能达到0.02~1.34μg/kg,这一结果与本方法较为接近。

表2 标准曲线线性方程、相关系数、检出限及线性范围Table 2 Linear regression equations,correlation coefficients,detection limits and linear ranges

a—调制周期为1s;b—调制周期为2s;c—调制周期为3s;d—调制周期为4s(峰号所对应的化合物名称见表2)。图1 24种混合标准溶液4种调制周期色谱图Fig.1 Gas chromatograms (GC×GC-μECD) of 24 mixed standard solution in 4 cycles
2.5.2方法精密度和回收率
选用实际空白土壤样品作为基质,分别添加1.0μg/kg和20.0μg/kg两个浓度水平混合标准溶液,按实验方法进行前处理,平行测定6次,计算回收率及测定值的相对标准偏差(RSD)。由表1中的数据可知,2个浓度水平的加标回收实验中,索氏提取法对各物质的回收率在80.7%~103.5%之间,ASE与索氏提取基本相当,仅个别物质回收率略低,超声波提取的回收率和精密度明显低于前两种方法。主要考虑以下两个因素:第一,本地区农田土壤含水率较大,尽管提取前加入足量无水硫酸钠混合分散,但超声时还是极易结块,从而影响回收率和精密度。第二,索氏提取使用了250mL索氏套筒,空间较大,一方面可添加足量无水硫酸钠混合分散,另一方面较大的套筒空间使得回流频率降低,增加了样品在溶剂中的浸泡时间,这使得索氏提取法的优势更加明显。这也可能是本方法回收率和精密度均优于马玲等[32]采用的超声波提取法的原因。
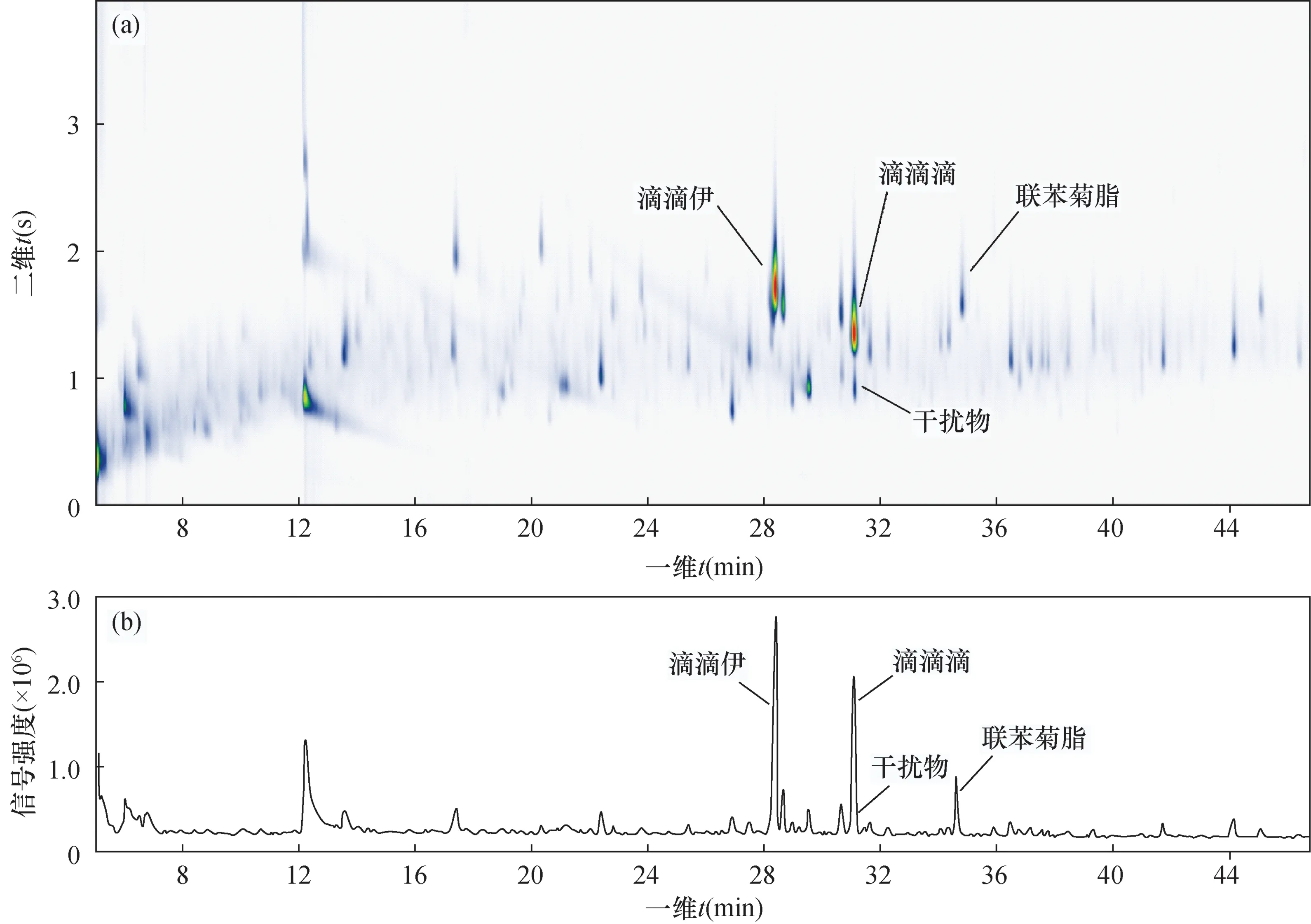
a—样品5全二维气相色谱图;b—样品5一维气相色谱图。图2 样品5的全二维色谱图与一维色谱图比较Fig.2 Comparison of GC×GC-μECD chromatragram and GC-μECD chromatragram of Sample No.5
2.6 实际样品测定
应用该方法测定2018年全国土壤详查江苏地区土壤样品56份,其中21份样品检出有机氯农药,检出物主要为滴滴伊、α-六六六、β-六六六和p,p’-滴滴涕,少量样品检出六氯苯,拟除虫菊酯类农药检出率较低,仅1份样品检出少量联苯菊酯。对比样品5的一维气相色谱图(图2b),可见全二维色谱法在二维图谱上成功分离了干扰物,保证了定量准确度(图2a),平行样的相对偏差低于25%,能够满足2018年全国土壤详查样品检测的质控要求,检测结果用GC-MS法进行验证(表3),同一样品检出物均一致,检出值的相对偏差在3.55%~23.08%之间,56份样品未出现1例假阳性结果。

表3 样品验证结果比对Table 3 Comparison of analytical results by GC×GC-μECD and GC-MS
3 结论
本工作建立了复杂基质土壤中24种有机氯和拟除虫菊酯类农药的全二维气相色谱检测方法,经实际样品的验证,证实该方法可有效改善基质干扰物和目标物的分离效果,准确度和灵敏度均较高,对于干扰物较多的样品具有一定实用性。
目前本方法只使用了一根二维色谱柱与三种一维柱进行优化,从色谱峰的分离效果来看仍有较大改进空间,后期将购买不同类型二维柱进行比较试验,针对高污染、多干扰的样品提供有效的解决方案。值得注意的是,尽管全二维气相色谱在一定程度上增加了峰容量,提高了分离度,但前期的净化仍至关重要,否则过多的干扰物会污染仪器,同时影响检测准确性。
猜你喜欢
石油炼制与化工(2022年2期)2022-02-15
海南热带海洋学院学报(2021年5期)2021-11-07
森林工程(2021年4期)2021-08-23
化工管理(2020年26期)2020-10-09
石油工业技术监督(2019年5期)2019-05-30
山东化工(2019年2期)2019-02-21
农产品加工(2018年9期)2018-05-25
资源节约与环保(2018年1期)2018-02-08
速读·中旬(2016年9期)2017-05-09
电子技术与软件工程(2016年24期)2017-02-23
