
百菌清降解菌的分离鉴定及功能基因分析
2020-12-25 01:24任晓洁贺壮壮赵玉斌宋元达赵新河
农业工程学报 2020年19期
任晓洁,贺壮壮,单 昕,赵玉斌,宋元达,赵新河,,4
百菌清降解菌的分离鉴定及功能基因分析
任晓洁1,2,贺壮壮1,单 昕1,赵玉斌3,宋元达1,赵新河1,3,4※
(1. 山东理工大学农业工程与食品科学学院,考林腊特列杰微生物脂质国际研究中心,淄博 255000;2. 保龄宝生物股份有限公司,德州 253000;3. 鲁洲生物科技有限公司,临沂 276400;4. 重庆市科学技术研究院,重庆 401123)
百菌清是一种广谱非内源性农药,在土壤中难以降解,已经成为农业环境污染的主要污染源之一。因此,其对环境的污染和被污染土壤的修复技术越来越受到关注。土壤环境中原位降解细菌的多样性对于评价环境毒理学、生物降解性、自净化能力和污染物的修复潜力具有重要价值。该研究从长期被百菌清污染的土壤中收集大量样本,分离到了14种能够明显降解百菌清的细菌。根据菌株形态和rDNA同源性分析,将它们分为假单胞菌属(sp)、无色杆菌属(sp)、苍白杆菌属(sp)、青枯菌属(sp)和溶杆菌属(sp)。其中溶杆菌属是该研究中新发现的具有百菌清降解能力的功能菌属,该发现扩大了已知的百菌清降解菌的菌属范围。通过进一步鉴定及生理生化分析,确定了该降解菌的分类地位及理化特征。此外,该研究通过基因文库法克隆到了发挥关键降解作用的水解酶基因,并发现该基因与转座子原件相连,二者组成代谢转座子,具有水平转移的分子基础。通过揭示降解基因在细菌间的漂移机制,初步明确了降解菌的功能基因及其分布规律。
土壤;农药;细菌;基因克隆;水平漂移
0 引 言
百菌清(2, 4, 5, 6-四氯间苯二甲腈)是一种非内吸性广谱杀菌剂,对多种作物真菌病害具有防治作用。该农药性质稳定,残效期长,具有明显的毒性蓄积。受到百菌清污染的农业环境对人们的生活产生重大影响,该农药对人和动物内分泌系统产生干扰作用,会造成人和动物雌性化、腺体病变和后代生命力退化,进而影响人和动物的生殖繁衍。因此,受百菌清污染土壤的修复技术也越来越受到人们的关注。农药进入土壤后会对土壤微生态环境产生一定的影响[1-3]。研究证明,百菌清会对不同的土壤微生物生长造成相应的刺激或者抑制[4-7]。冯波等[8]在模拟土壤生态系统中研究了百菌清对土壤微生物数量和酶活性的影响。研究后发现,土壤经不同浓度百菌清处理后细菌、放线菌和真菌的生长受到不同程度的抑制作用。Sigler等[9]也对百菌清污染的土壤微生物群落进行分析,发现百菌清能够抑制或者刺激不同细菌的生长,而真菌则全部被明显抑制。在对土壤中生物酶的研究发现,百菌清对土壤酸性磷酸酶、碱性磷酸酶、脲酶、蔗糖酶、过氧化氢酶、纤维素分解酶均能够产生一定的抑制作用[4,8,10]。因此,百菌清被广泛证实能够从多方面影响土壤微生态环境。
利用特种微生物降解土壤中的农药残留是农业土壤修复的重要手段之一。某些微生物可以将有机农药污染物作为原料进行分解,从而达到降解的目的[11-12]。国内外在利用微生物降解农药的研究中已经发现了大量的有机农药降解菌,比如,人们分离到了众多能够分解对硫磷、甲基对硫磷及其他有机磷的生物降解菌[13-15]。然而,在百菌清的生物降解研究中,只有少数降解菌得到了研究[16-18]。并且,在研究百菌清生物降解菌的过程中,其降解功能基因的分离及分析等工作十分欠缺。在其他农药降解菌的降解机理研究中发现,降解菌的生物多样性与分子水平漂移机制相关。例如,研究发现降解有机磷基因的DNA中鸟嘌呤与胞嘧啶碱基对摩尔百分数含量与降解菌自身染色体DNA中的鸟嘌呤与胞嘧啶碱基对摩尔百分数含量明显不同,降解基因如同一个外源侵入基因,插入到降解菌的DNA中,从而使其获得了降解污染物的能力。此外,进化上相近的降解基因从发育关系较远的微生物中被发现[19-21],同样也被认为是降解基因通过水平转移获得[22]。目前,已有很多研究从诸多方面揭示了降解基因水平漂移的现象[23],并证明发生基因的水平漂移,是微生物获得降解功能的重要途径。并且,已有大量研究获得了多种降解农药的代谢转座子。例如,Siddavattam等[24]发现有机磷降解基因存在于一个类转座子结构中;张瑞福等[25]发现7株菌中甲基对硫磷基因都与转座元件IS6100紧密相连;Hoffmann[26]发现2,4-D降解菌中降解基因的基因簇两侧有IS1071和IS1380两个转座元件,组成一个30 kb的定位于染色体上的转座子结构。Top等[27-28]也研究了2,4-D降解质粒在不同降解菌中的转移作用及其对土壤中2,4-D降解的影响。然而,百菌清降解基因的代谢转座元件尚无报道。
因此,研究百菌清的生物降解,丰富降解菌库,研究百菌清降解菌的多样性,并从基因水平克隆和分析发挥关键降解作用的基因等,能够为今后受百菌清污染土壤的生物修复技术奠定理论基础。本文通过百菌清降解菌的分离和鉴定,研究了百菌清降解菌的多样性,并对百菌清降解基因及其在各菌属中的散布机制进行了初步研究,为受百菌清污染土壤的生物修复技术研究奠定了基础。
1 材料与方法
1.1 降解百菌清菌株的分离
分别从百菌清生产车间、生产车间的绿化带和连续施用百菌清的农田表层(0~15 cm)收集土壤样品。分离所用的筛选培养基有基础矿物盐培养基(Minimal salt medium,MSM,普遍适用于各类微生物的筛选)、溶菌肉汤培养基(Lysogeny broth,LB,特别适用于细菌的筛选),马铃薯葡萄糖琼脂培养基(Potato Dextrose Agar,PDA,特别适用于放线菌的筛选)及高氏一号培养基(特别适用于真菌的筛选)。MSM培养基每升包含1.0 g NH4NO3、0.5 g MgSO4·7H2O、0.5 g (NH4)2SO4、0.5 g KH2PO4、0.5 g NaCl和1.5 g K2HPO4,pH值7.0。LB培养基每升含10.0 g胰蛋白、5.0 g酵母提取物和10.0 g NaCl(pH值7.0)。PDA培养基包含200.0 g马铃薯和20.0 g蔗糖(pH值7.0)。高氏一号培养基中含1.0 g KNO3、0.01 g FeSO4·7H2O、0.5 g K2HPO4、0.5 g NaCl、20.0 g淀粉(pH值7.2)。固体培养基的配制通过添加2.0%琼脂来制备。各种培养基在121℃高压灭菌20 min,将百菌清储备液过滤除菌并添加到各种筛选培养基中,以使最终浓度达到5、10、15、20、25、30、35、40、45、50、60、80、100 mg/L。全部试剂均为分析纯。
采用稀释平板法分离百菌清降解菌:将1 g土壤放入9 mL无菌水中,并在30 ℃下混合20 min。然后静置30 min后,取出1 mL土壤悬浮液,并稀释至其原始浓度的10-3、10-4、10-5。然后将每种浓度的200L稀释液涂在含有百菌清的初筛培养基MSM上。在30 ℃下孵育2~7 d后,根据菌落周围的水解环选择分离株。挑选产生透明圈的单个菌落,在筛选培养基上划线多次,并在含有百菌清的筛选培养基上重新检查纯化的菌株。
1.2 百菌清降解菌株的鉴定与表征
根据伯杰氏细菌学手册,结合菌株16S rDNA及生理生化特性对其进行鉴定。根据标准方法提取基因组DNA。通过使用2个通用引物P1和P6,从其基因组DNA扩增16S rRNA。其中,正向引物P1(59-AGAGTTTGATCCTGGTCAGAACGCT-39)对应于大肠杆菌16S rRNA基因的8-37位置,反向引物P6(59-TACGGCTACCTTGTTACGACTTCACCCC-39)对应于大肠杆菌16S rRNA基因的1479-1506位置。聚合酶链式反应(polymerase chain reaction,PCR)条件为:94 ℃预变性5 min,变性(94 ℃,30 s)、退火(55 ℃,30 s)、延伸(72 ℃,90 s)作为一个循环,共进行30个循环。最后在72 ℃稳定延伸10 min。PCR产物用凝胶提取试剂盒(TaKaRa Bio, Otsu, Japan)纯化,并由中国北京的三博生物技术有限公司进行测序。应用NCBI中的BLAST进行DNA序列分析。从Genbank中调取亲缘关系近的菌株的16S rDNA序列,用MEGA3.1进行序列比对,计算遗传距离,建立系统发育树。
用于菌种鉴定的生理生化实验包括革兰氏染色,氧化酶,接触酶,最适pH值,耐盐性试验,硝酸还原试验,酪蛋白水解试验,产吲哚试验,七叶苷水解试验,尿素水解,吐温80水解,淀粉水解,醌、极性脂、脂肪酸含量测定等,各生化反应的具体方法参照[29]。
1.3 染色体DNA中鸟嘌呤和胞嘧啶碱基对的摩尔含量和DNA的同源性测定
根据细菌分类鉴定标准,细菌染色体DNA中鸟嘌呤和胞嘧啶碱基对的摩尔含量及染色体DNA的同源性是将细菌鉴定到种的依据。鸟嘌呤和胞嘧啶碱基对的摩尔含量测定采用热变性温度(T值)法,所用仪器由Lambda Bio 20型紫外分光光度计、PTP-1 温度控制仪、循环水浴和计算机组成。整个测定过程由UV Winlab 软件控制。
1)T值的测定:将待测DNA样品适当稀释,使其吸光度为0.2~0.5。以K12菌株的DNA作参比,以消除试验系统误差。将稀释后的DNA置于带加热装置的紫外分光光度计中完成热变性,根据变性过程中各温度和对应的相对吸光度绘制成DNA变性曲线,热变性曲线中点的温度即为T值。
2)所测T值带入特定公式即可算出细菌染色体DNA的鸟嘌呤和胞嘧啶碱基对的摩尔含量。使用公式鸟嘌呤和胞嘧啶碱基对的摩尔含量= 51.2+2.08 [T()−T()]计算,其中T()为待测菌的T值;T()为大肠杆菌K12的T值。
细菌染色体DNA同源性的测定采用DNA-DNA杂交法,经过DNA提取,剪切,变性,复性,从而测定两株菌的DNA复性速率和同源性。将DNA置于温度控制仪,并在100℃下变性10 min,变性结束后,将温度控制仪的温度设定为最适复性温度[T=0.51(鸟嘌呤和胞嘧啶碱基对的摩尔含量)+ 47.0]。当变性DNA样品的温度迅速降至最适复性温度,并稳定2 min后,开始复性反应,一般进行20 min。计算机可记录260 nm处吸光值随时间变化的复性反应曲线,该曲线的斜率即为DNA 样品的复性速率(通常用表示,单位为每分钟吸光值的减少值)。DNA同源性(,%)根据公式计算:
=[4V−(V+V]/[2(V∙V1/2]×100 (1)
式中V表示样品A的自身复性速率,V表示样品B的自身复性速率,V表示样品a与b等量混合后的复性速率。
1.4 基因文库构建及功能基因筛选
提取溶杆菌RB-38的基因组DNA,取4g DNA,加入2L稀释至 0.03 U/L的Sau3AI酶液,37℃消化,反应35 min,琼脂糖电泳,回收4-8 kb片段。由于Sau3A I与BamH I为同尾酶,所以采用内切酶BamH I对载体pUC19进行酶切,并用碱性磷酸酶(CIAP)处理。将酶切片段与酶切的pUC19载体连接,连接体系为10L,载体0.5L,外源8.2L,10×buffer 1L,连接酶0.3L,16 ℃过夜连接,然后取连接产物1L转化大肠杆菌DH10B,电极槽为1 mm,电压1 600 V。将基因文库得到的转化子在百菌清液体筛选培养基中连续培养三代,最终收集菌体点种于百菌清筛选固体培养基上,观察透明圈产生情况,从而筛选具有百菌清降解功能的基因片段。
2 结果与讨论
2.1 百菌清降解菌的分离与鉴定
不同微生物对百菌清的耐受程度是不一样的,为了选择合适的培养浓度,保证降解菌不被过高浓度的农药抑制或杀死,在各筛选培养基中加入百菌清母液,制成农药梯度培养基,并观察结果,见表1。结果显示细菌对百菌清的耐受性最强,其在加有100g/mL百菌清的LB细菌培养基上的仍能大量存活,而真菌在添加同样浓度百菌清的高氏一号培养基上不生长。百菌清浓度超过60g/mL,用于分离真菌的高氏一号培养基上不出现菌落,而百菌清浓度低于40g/mL,用于分离细菌的LB平板上的菌落数过多,不利于后续操作。因此选择百菌清终浓度为50g/mL的培养基进行后续的培养。
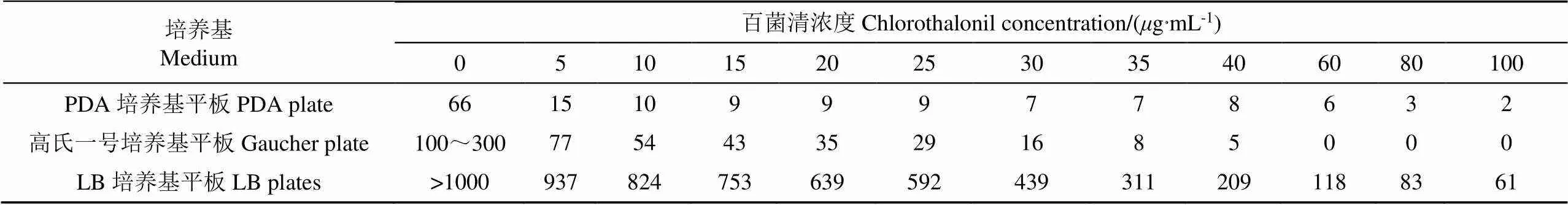
表1 含不同浓度百菌清的培养基上菌落个数
由于待筛选的土壤中微生物的类型未知,因此我们初始选用适宜各种微生物生长的微生物基础培养基(基础矿物盐MSM培养基),加入50g/mL百菌清进行筛选。本试验共分离纯化出14株百菌清降解菌,它们在含有百菌清的MSM平板上均有明显的透明圈产生,见图1。由培养结果可以看出,大部分筛选到的菌株可以在培养2 d之后就对百菌清产生明显的降解作用,因此具有非常好的应用潜力。
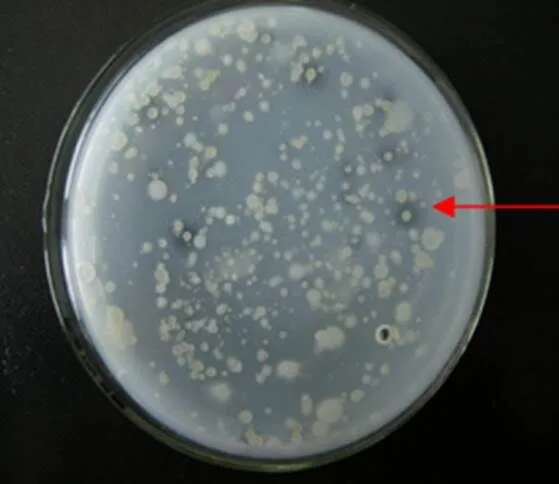
注:筛选平板为加有50 μg∙mL-1百菌清的基础矿物盐MSM培养基,箭头指示在菌落周围产生降解圈的菌,为筛选到的具有百菌清降解功能的菌株。
经革兰氏染色,这些降解菌均为革兰氏阴性菌,将这些分离到的菌株在含有百菌清的LB培养基上点种,验证其百菌清降解功能(图2),这14株菌均有明显的降解百菌清的能力。显微镜下观察菌体的形态各有不同(表2)且均无芽孢。

注:RB-No. 为各降解菌株在筛选过程中的编号,下同。
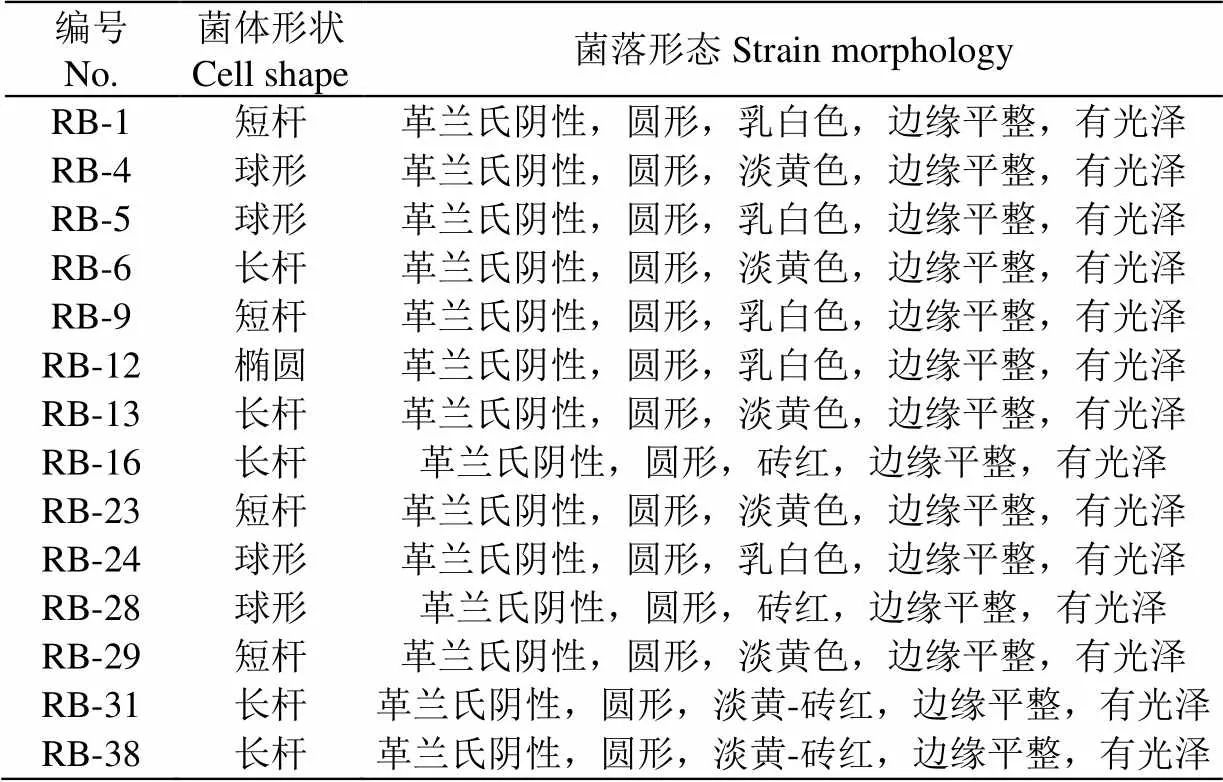
表2 百菌清降解菌的表型特征
扩增各降解菌株的16SrDNA,PCR产物纯化回收后连接pMD18-T载体,转化大肠杆菌感受态细胞DH5α,挑取在Amp-X-gal-IPTG上的白斑,cracking检测后,送北京三博生物技术有限公司测序,测序结果在NCBI上用BLAST在线分析,结果显示菌株RB-1、RB-4、RB-5、RB-6、RB-9、RB-12、RB-13、RB-23、RB-24的16S rDNA序列与假单胞菌属()的多株菌同源性达99%,菌株RB-16与无色菌属()的多株菌同源性达99%,菌株RB-28与苍白杆菌属()多株菌同源性高达99%,菌株RB-29与青枯菌属()多株菌同源性达99%,菌株RB-31与RB-38与溶杆菌属()多株菌同源性达97%。通过分析比较将14株菌初步鉴定到属的水平,如表3所示,并构建了它们之间16S rDNA序列同源性系统发育树(图3)。本研究表明百菌清降解菌种类广泛,并以假单胞菌的数量和种类最多。在过去的研究中分离到的百菌清降解菌有氮单胞菌属(),黄杆菌属(),莫拉氏菌属(),假单胞菌属(),微球菌属()和苍白杆菌属()等[16-18]。本试验分离到的假单胞菌属和苍白杆菌属为已知的具有百菌清降解功能的菌属,而无色菌属、青枯菌属及溶杆菌属是最新分离到的具有百菌清降解功能的菌株。这说明百菌清对土壤微生物种类的影响有可能还跟土壤类型、土壤性质、土壤中土著微生物的分布及丰度等因素有关。本试验的筛选在前人研究的基础上进一步丰富了百菌清降解菌库,增加了无色菌属、青枯菌属及溶杆菌属的降解菌株。此外,本试验筛选到的溶杆菌属的RB-31、RB-38与该属内其他标准菌株的同源性较低,16S rDNA序列同源性只有97%,该值处于细菌新种鉴定的临界值,说明其有可能成为该菌属中的新种,因此选定这2株菌进行进一步的鉴定。

表3 百菌清降解菌的16SrDNA同源性比对结果

图3 百菌清降解菌的16S rDNA系统发育树
用MEGA3.1软件分别计算RB-31、RB-38与溶杆菌属17株标准菌株的16S rDNA的遗传距离,构建系统进化树(图4),结果表明RB-31和RB-38位于同一个分支,且与标准菌株GH1-9T的进化关系最近,同源性分别为97.4%和97.2%。与其他标准菌株的同源性都低于97%。16S rDNA 序列同源性低于97%的菌一定不是同一个种,而具有97%或以上的相似性时,不能判定是哪个种,需要进一步DNA-DNA杂交确定,因此要确定RB-31和RB-38是不是溶杆菌属的新种需要将其分别于GH1-9T进行DNA-DNA杂交。结果表明,RB-31与GH1-9T的DNA同源性为49.9%,RB-38与GH1-9T的DNA同源性为20.07%,RB-31与RB-38之间的DNA同源性超过70%,根据国际系统细菌学委员会规定,DNA同源性≥70%,杂交分子的热解链温度差≤5℃为细菌种的最低界限[30]。由于两株菌与标准菌株GH1-9TDNA之间同源性均小于70%,而两株菌之间DNA同源性超过70%,所以可以初步确定RB-31、RB-38代表溶杆菌属的一个新种,为同一种内的不同菌株。
2.2 分离菌株的生理生化特性
由于生理生化特征往往是菌株本身某种代谢途径或某种酶的特有表现,它在某种程度上反映了菌株的本质特征,因此本研究选取有代表性的菌株RB-38进行生化性质的测定,测定结果如表4。由前面结果已知,RB-38经过16S rDNA测序被初步鉴定为溶杆菌属,其与该属内其他标准菌株的同源性较低(97%左右),且与标准菌株GH1-9T的进化关系最近,同源性为97.2%。因此,将其与标准菌株GH1-9T进行生理生化特征的比较,结果表明RB-38与参比菌株GH1-9T生理生化特征相似,进一步证明RB-38是溶杆菌属的菌株。然而同一属中不同种之间的菌株性状也有个别不同之处(表4)。其中,RB-38较GH1-9T耐受的温度范围较窄,但它的耐酸碱能力和耐盐能力范围均比GH1-9T广,这一突出的酸碱适应性使得RB-38菌株在处理土壤及农药污染方面具有了更大的应用潜质。
2.3 百菌清降解基因的克隆及分布
将基因文库得到的一万多株转化子在百菌清液体筛选培养基中连续培养三代,最终收集菌体点种于百菌清筛选固体培养基上,观察透明圈产生情况,从而筛选具有百菌清降解功能的基因片段。筛选到的具有百菌清降解功能的阳性转化子编号838,提取阳性转化子838的质粒pU838再次转化大肠杆菌进行功能验证,效果如图5。
对阳性质粒pU838测序分析,结果表明其插入片段为3 493 bp,通过NCBI BlastX和软件DNAMAN5.2中的ORF finder的分析表明,这段序列上有3个ORF,且转录方向一致。其中ORF1编码转座酶,ORF2编码ATP结合蛋白,ORF3编码水解脱卤酶。
经与NCBI数据库中的已知序列进行Blast比对,ORF3编码的氨基酸序列与sp. CTN-3中水解脱卤酶氨基酸序列一致性达99%。推测发挥百菌清降解功能的应该为此水解脱卤酶(),推测百菌清在该酶的作用下苯环上的氯原子被水解,从而达到降解目的。
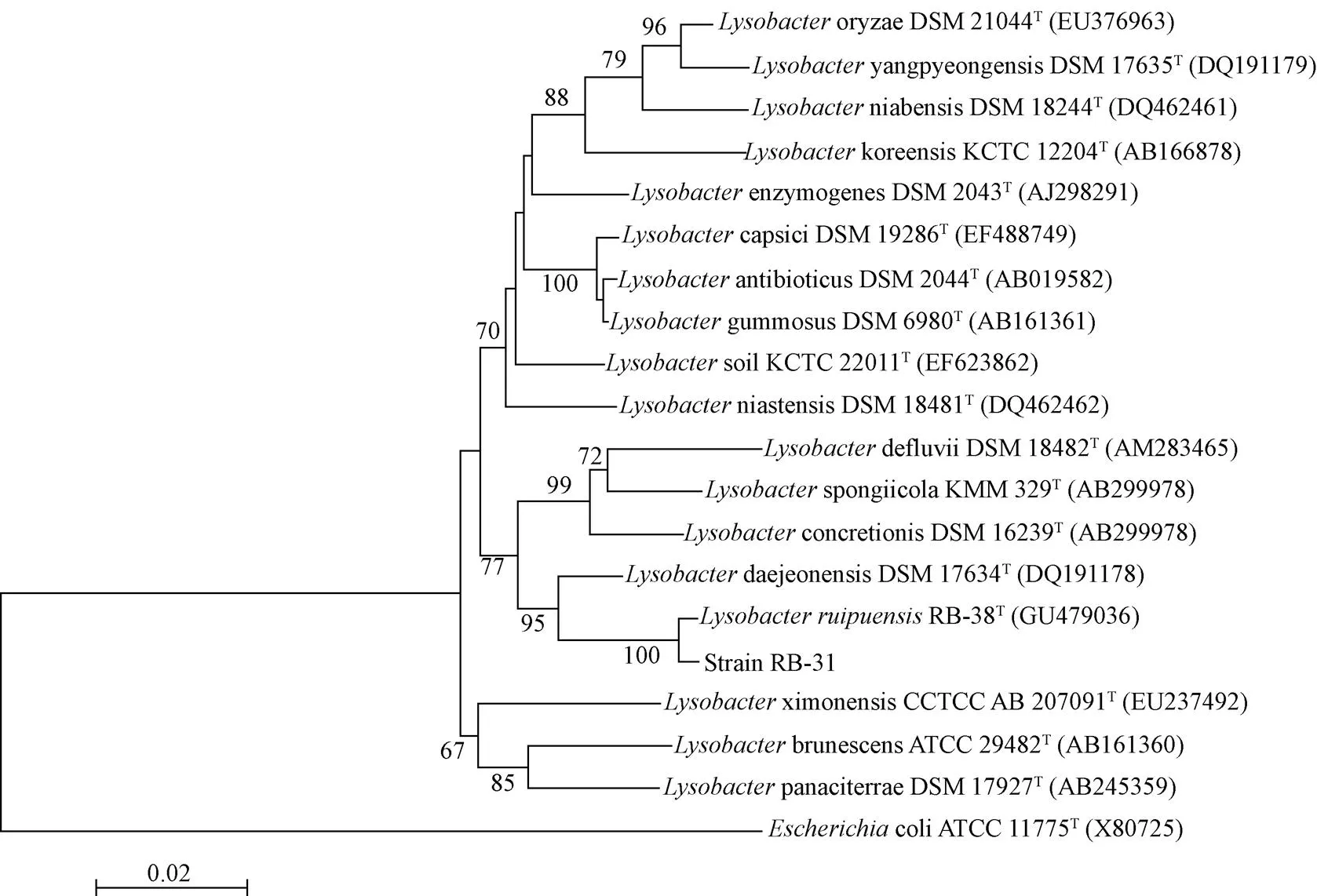
注:Escherichia coil ATCC 11775T为组外参照。

表4 菌株RB-38和标准菌株L.daejeonensis GH1-9T特征对比
注:GH1-9T为标准菌株 (standard strain);+:阳性反应; -:阴性反应;W:微弱生长。
Note:GH1-9Tis standard strain; +: positive reaction; -: negative reaction; W: weak growth.
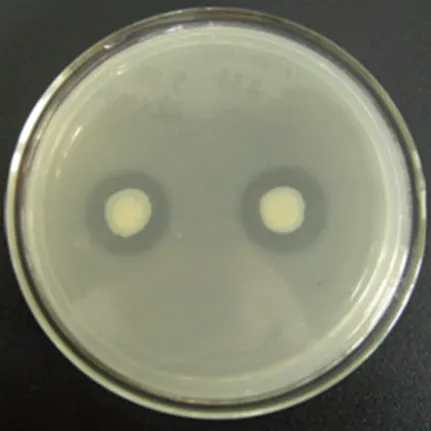
图5 阳性转化子质粒pU838在大肠杆菌中的功能验证
针对ORF3的序列,设计特异引物,扩增结构基因,5′端引物P1(5′-AGAAAGCTTGACG ATGCCACTC-3′),引了I酶切位点,3′端引物P2 (5′-AAGTCTAGAGAGCAGGATCAAG GC-3′),引入了d III酶切位点,扩增产物与经过相同酶切后的pUC19连接,使得结构基因的起始密码子在pUC19的启动子下融合表达,验证该阅读框区域的功能,构建的载体命名为pUchd。结果如图6所示:大肠杆菌coil DH10B转化子产生了降解百菌清的透明圈,证明该开放阅读框所编码的水解脱卤酶正是降解百菌清的功能酶,且该酶的表达不需要其他调控序列的存在。
进一步对3个开放阅读框之间的核苷酸序列分析表明在ORF1和ORF2两侧存在20 bp的反向重复序列,其中左侧重复序列(IRL)为5′- AAAACTGGGCCACTTCGCGC-3′,右侧重复序列(IRR)为5′-GCGCGAAGTGGCTCAGATTT-3′。这4个元件(IRL、ORF1、ORF2、IRR)之间的组成和排列关系与已知的IS21家族简单插入序列[31]具有典型的相似性,该家族插入元件均含有转座酶编码区、ATP-结合蛋白编码区,并被2个简单的反向重复序列所包围。IS21家族中,研究的比较多的是IS100,IS100的结构特征于1994年就被Alexander等研究人员研究清楚,它全长1 954 bp,两端各有28 bp的反向重复序列,一个1 023 bp的转座酶编码区和一个783 bp的istB样的ATP-结合蛋白的编码区[32]。因此本研究在此也得到了一个IS21家族的新成员,定名其为IS-,其全长1 926 bp,包含一个1 023 bp的转座酶编码区和一个657 bp的ATP-结合蛋白编码区,两端各有20 bp的反向重复序列。

注:2个箭头均代表阳性转化子E.coli DH101/pUchd。
为探索不同的百菌清降解菌中发挥降解作用的基因之间异同,本研究根据RB-38中已得到的序列设计特异性引物,引物在序列中的位置及关系如图7所示:P1,P2用于扩增结构基因,838上游P1,P2用于扩增及上游序列。扩增结果如图8所示,供试菌中均可扩增到与阳性对照大小完全相同的条带,选取部分阳性条带测序发现均为基因,然而并不是所有降解菌中都可以扩增到与基因相连的上游序列,扩增到上游序列的菌株有RB-4、RB-5、RB-13、RB-23、RB-24、RB-29,分别属于假单胞菌属和青枯菌属。本研究分析发现,多株菌中百菌清降解酶基因均与简单插入序列IS-相连,组成一个代谢转座子,因此推测该转座子是百菌清降解酶基因水平转移的分子基础,百菌清降解基因在IS-的作用下水平转移,并出现在多株降解菌中。

图7 特异性引物的相对位置及关系示意图
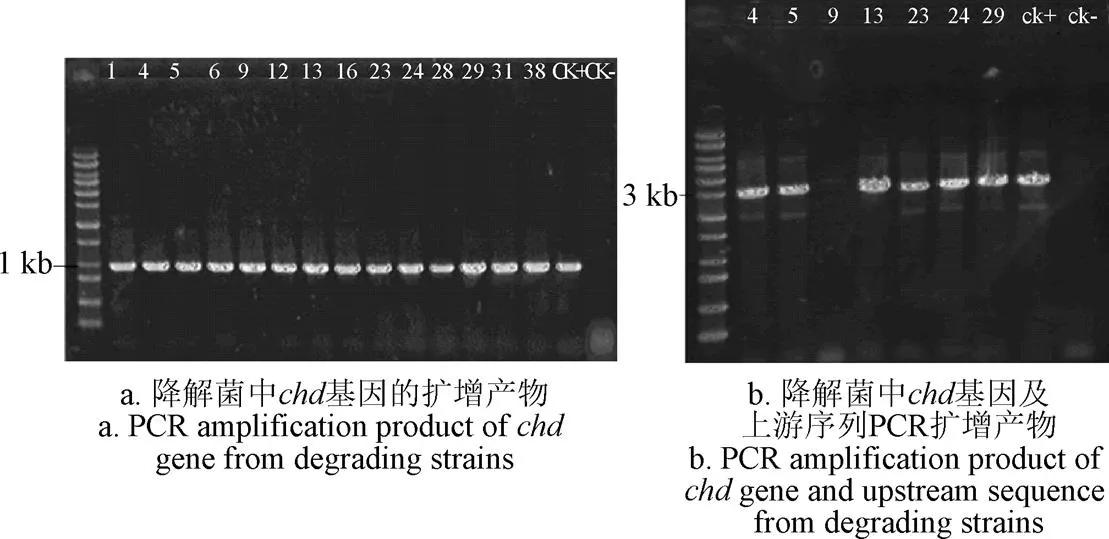
注:数字对应于相应的菌株序号RB-(N); Marker; 1kb plus DNA ladder
目前分离到的有机物降解菌分类广泛,包括假单胞菌属()、无色杆菌属()、产碱菌株()、屎拟杆菌()、吉氏拟杆菌()、芽孢杆菌属()、棒状杆菌属()、土壤杆菌属()、黄杆菌属()、短杆菌属()、枝动杆菌属()、黄孢原平革菌属()、曲霉属()、青霉属()、根霉属()、镰刀菌属()、总状共头霉()、诺卡氏菌属()、链霉属()等[11]。然而农药降解基因之间的保守性却很高,南京农业大学李顺鹏实验室分离到的7株甲基对硫磷降解菌分别为假单胞菌属()、无色杆菌属()、布鲁氏菌()和苍白杆菌属(),然而他们发挥降解作用的基因却都是甲基对硫磷降解酶基因methyl parathion-degrading(gene)[12]。国内外分离到了很多对硫磷、甲基对硫磷及其他有机磷降解菌,他们分别属于黄质菌属(),假单胞菌属(),无色杆菌属()[13-15],然而这些菌都合成有机磷农药水解酶基因organophosphorus pesticide degrading () gene,通过有机磷农药水解酶发生降解作用[33]。其中假单胞菌GM和黄质杆菌sp. strain ATCC 27551中的基因完全相同,并与黄质杆菌中该基因的同源性为98%。有机污染环境中污染物降解菌的多样性及降解基因的保守性,揭示了污染环境中微生物之间也许存在着降解基因的交流。
目前已经确定了3种基本的介导细菌间水平基因转移的机制。细菌接合是依赖于细胞接触的特定质粒或转座子从供体到受体细胞转移。自然转化是在特定条件下细胞产生感受态并吸收自由DNA的过程。转导是噬菌体介导的基因信息在供体和受体细胞的转移。一些试验现象和证据将研究者的思路引向了基因水平漂移的领域。首先人们发现进化上相近的降解基因或基因簇其宿主菌的地理位置上很远[21]。而同一污染物不同菌株降解基因的系统进化关系与宿主菌16SrDNA的系统发育关系不一致。接着人们又发现降解有机污染物的基因与自转移质粒或转座子等移动基因元素相连:Siddavattam等[24]发现有机磷降解基因存在于一个类转座子结构中,张瑞福等[25]发现7株菌中甲基对硫磷基因都与简单插入序列IS6100紧密相连,同时降解基因或基因簇鸟嘌呤和胞嘧啶碱基对的摩尔含量与宿主菌的明显不同,Hoffmann等[26]发现2,4-D降解菌P4a中降解基因的基因簇两侧有IS1071和IS1380两个简单插入序列,组成一个30 kb(Kilobase,千碱基)的定位于染色体上的转座子结构。Top等[28]也研究了2,4-D降解质粒在不同降解菌中的转移作用及其对土壤中2,4-D降解的影响。因此宿主菌降解基因的水平转移是造成降解菌多样性及其环境适应能力产生的很重要途径。
3 结 论
本研究从百菌清生产车间,生产车间的绿化带和连续施用百菌清的农田表层采集土样,利用稀释平板法,在含有百菌清的平板上分离到14株百菌清降解菌,经16S rDNA系统发育分析将所有菌株鉴定到假单胞菌属()、无色杆菌属()、苍白杆菌属()、青枯菌属()和溶杆菌属(),并经过进一步的鸟嘌呤和胞嘧啶碱基对的摩尔含量的测定、DNA-DNA杂交、化学鉴定及生理生化特征的鉴定最终将其中的2株鉴定到种的水平,为后续的工作奠定了基础。同时构建了百菌清降解菌的基因组文库,并从中克隆到了降解基因,初探了降解基因在污染环境中的多个菌株间水平转移的分子基础。本文的研究证明在百菌清污染的土壤中,微生物群落适应污染的分子机制之一是百菌清降解基因在细菌间的水平转移,降解基因的转移使得部分细菌获得了降解百菌清的能力,这是环境中百菌清降解菌多样性产生的原因。然而并不是所有的降解菌降解能力的产生都是通过这种方式得到的,部分降解菌中虽然也有同样的百菌清降解基因,但它们并没有处在转座元件的下游,这些基因有可能是降解菌本身就有的。因此对于该降解基因的起源还有待于进一步的研究。
[1] Chu H, Gao G F, Ma Y, et al. Soil microbial biogeography in a changing world: Recent advances and future perspectives[J]. mSystems, 2020, 5(2): 1-12.
[2] Min H, Ye Y F, Chen Z Y, et al. Effects of butachlor on microbial populations and enzyme activities in paddy soil[J]. Journal of Environmental Sciences, 2001, 365: 581-595.
[3] Haque M N, Eom H J, Nam S E, et al. Chlorothalonil induces oxidative stress and reduces enzymatic activities of Na+/K+-ATPase and acetylcholinesterase in gill tissues of marine bivalves [J]. PLoS One. 2019, 14(4): 1-17.
[4] Baćmaga M, Wyszkowska J and Kucharski J. The influence of chlorothalonil on the activity of soil microorganisms and enzymes[J]. Ecotoxicology, 2018, 27(9): 1188-1202.
[5] Teng Y, Zhang M, Yang G, et al. Successive chlorothalonil applications inhibit soil nitrification and discrepantly affect abundances of functional genes in soil nitrogen cycling [J]. Environmental Science and Pollution Research International. 2017, 24(4): 3562-3571.
[6] Wu X, Cheng L, Cao Z, et al. Accumulation of chlorothalonil successively applied to soil and its effect on microbial activity in soil[J]. Ecotoxicology and Environmental Safety. 2012, 81: 65-69.
[7] Devi Y B, Thounaojam Meetei T, Kumari N. Impact of pesticides on soil microbial diversity and enzymes: A review[J]. International Journal of Current Microbiology and Applied Sciences. 2018, 7(6): 952-958.
[8] 冯波,单敏,方华,等. 百菌清对土壤微生物数量和酶活性的影响[J]. 农业环境科学学报,2006(3):674-677.
Feng Bo, Shan Min, Fang Hua, et al. Effects of chlorothalonil on soil microbial populations and enzyme activities[J]. Journal of Agro-Environment Science, 2006(3): 674-677. (in Chinese with English abstract)
[9] Sigler W V, Turco R F. The impact of chlorothalonil application on soil bacterial and fungal populations as assessed by denaturing gradient gel electrophoresis[J]. Applied Soil Ecology, 2002, 21(2): 107-118.
[10] Yang X, Bennett B, Holz R C. Insights into the catalytic mechanism of a bacterial hydrolytic dehalogenase that degrades the fungicide chlorothalonil[J]. Journal of Biological Chemistry, 2019, 294(36): 13411-13420.
[11] 史秀珍. 百菌清降解菌的筛选及其降解特性研究[D]. 北京,中国农业科学院,2007.
Shi Xiuzhen. Isolation and Characterization of A Chlorothalonil-Degrading Bacterium[D]. Beijing: Chinese Academy of Agricultural Sciences, 2007. (in Chinese with English abstract)
[12] Zhang R, Cui Z, Zhang X, et al. Cloning of the organophosphorus pesticide hydrolase gene clusters of seven degradative bacteria isolated from a methyl parathion contaminated site and evidence of their horizontal gene transfer[J]. Biodegradation, 2006, 17(5): 465-472.
[13] Ogbo F C. Conversion of cassava wastes for biofertilizer production using phosphate solubilizing fungi[J]. Bioresource Technology, 2010, 101: 4120-4124.
[14] Patel D K, Murawala P, Archana G, et al. Repression of mineral phosphate solubilizing phenotype in the presence of weak organic acids in plant growth promoting fluorescent[J]. Bioresource Technology, 2011, 102: 3055-3061.
[15] Horne I, Sutherland T D, Rebecca L, et al. Identification of an(organophosphate degradation) gene in an agrobacterium isolate[J]. Applied and Environmental Microbiology, 2002, 68: 3371-3376.
[16] Liang B, Wang G, Zhao Y, et al. Facilitation of bacterial adaptation to chlorothalonil-contaminated sites by horizontal transfer of the chlorothalonil hydrolytic dehalogenase gene[J]. Applied and Environmental Microbiology, 2011, 77(12): 4268-4272.
[17] Liang B, Li R, Jiang D, et al. Hydrolytic dechlorination of chlorothalonil by. CTN-11 isolated from a chlorothalonil-contaminated soil[J]. Current Microbiology, 2010, 61(3): 226-233.
[18] Atashgahi S, Liebensteiner M G, Janssen D B, et al. Microbial synthesis and transformation of inorganic and organic chlorine compounds[J]. Frontiers in Microbiology, 2018, 9: 1-21.
[19] Whyte L G, Smits T H, Labbe M D, et al. Gene cloning and characterization of multiple alkane hydroxylase systems inQ15 and NRRLB-16531[J]. Applied and Environmental Microbiology, 2002, 68: 5933-5942.
[20] Chu H Y, Sprouffske K, Wagner A. Assessing the benefits of horizontal gene transfer by laboratory evolution and genome sequencing[J]. BMC Evolutionary Biology, 2018, 18(1): 1-21.
[21] Nielsen T K, Rasmussen M, Demanèche S, et al. Evolution of sphingomonad gene clusters related to pesticide catabolism revealed by genome sequence and mobilomics ofMH[J]. Genome Biology and Evolution. 2017, 9(9): 2477-2490.
[22] Muturi E J, Donthu R K, Fields C J, et al. Effect of pesticides on microbial communities in container aquatic habitats [J]. Scientific Reports, 2017(7): 1-10.
[23] Imperato V, Portillo-Estrada M, McAmmond B M, et al. Genomic diversity of two hydrocarbon-degrading and plant growth-promotingspecies isolated from the oil field of Bóbrka (Poland) [J]. Genes, 2019, 10(6): 1-22.
[24] Siddavattam D, Khajamohiddin S, Manavathi B, et al. Transposon-like organization of the plasmid-borne organophosphate degradation (opd) gene cluster found insp[J]. Applied and Environmental Microbiology, 2003, 69: 2533-2539.
[25] 张瑞福,戴青华,何健,等. 七株有机磷农药降解菌的降解特性比较[J]. 中国环境科学,2004(5):584-587.
Zhang Ruifu, Dai Qinghu, He Jian, et al. Comparison of degrading characteristics of seven organophosphate pesticide-degrading bacteria[J]. China Environmental Science, 2004(5): 584-587. (in Chinese with English abstract)
[26] Hoffmann D. A transposon encoding the complete 2, 4-dichlorophenoxyacetic acid degradation pathway in the alkalitolerant strainP4a[J]. Microbiology, 2003, 149(9): 2545-2556.
[27] Top T, Courde L, McGowan C, et al. Phylogenetic analyses indicate independent recruitment of diverse gene cassettes during assemblage of the 2, 4-D catabolic pathway[J]. FEMS Microbiology Ecology, 1999, 28: 373-382.
[28] Top E M, Springael D, Boon N. Catabolic mobile genetic elements and their potential use in bioaugmentation of polluted soils and waters[J]. Fems Microbiology Ecology, 2002, 42(2): 199-208.
[29] Jin H, Zhou Y, Liu H, et al. Paenibacillus jilunlii sp. nov., a nitrogen-fixing species isolated from the rhizosphere of Begonia semperflorens[J]. International Journal of Systematic and Evolutionary Microbiology 2011, 61: 1350-1355.
[30] Hinchliff C E, Smith S A, Allman J F, et al. Synthesis of phylogeny and taxonomy into a comprehensive tree of life[J]. Proceedings of the National Academy of Ences of the United States of America, 2015, 112(41): 12764-12769.
[31] Kim B J, Kim K, Kim B R, et al. Identification of ISMyo2, a novel insertion sequence element of IS21 family and its diagnostic potential for detection of Mycobacterium yongonense[J]. BMC Genomics, 2015, 16: 1-9.
[32] Lima-Mendez G, Oliveira A D, Ross K, et al. Toxin-antitoxin gene pairs found in Tn3 family transposons appear to be an integral part of the transposition module[J]. mBio, 2020, 11(2): 1-19.
[33] Trinder M, McDowell T W, Daisley B A, et al.reduces organophosphate pesticide absorption and toxicity to drosophila melanogaster[J]. Applied and Environmental Microbiology, 2016, 82(20): 6204-6213.
Isolation, identification and functional gene analysis of chlorothalonil degrading bacteria
Ren Xiaojie1,2, He Zhuangzhuang1, Shan Xin1, Zhao Yubin3, Song Yuanda1, Zhao Xinhe1,3,4※
(1255000,; 2...,253000,;3...,276400,; 4.,401123,)
Chlorothalonil (2, 4, 5, 6-tetrachloroisophthalonitrile, TPN) was used as a broad-spectrum and non-systemic fungicide in China. However, this pesticide has been classified as a “probable human carcinogen” by the U.S. Environment Protection Agency (US EPA), due to its highly toxic to birds, fish, and aquatic invertebrates. Alternatively, bioremediation can be expected to degrade, even remove organic pollutants, with the promising application prospects. The diversity of in situ degrading bacteria in a polluted environment is critical to evaluate environmental toxicology, biodegradability, self-purification ability, and remediation potential of pollutants. In this study, an attempt was made to apply the biodegradation for the control of pollution. Firstly, the soil samples were collected from the long-term chlorothalonil-contaminated field. Fourteen chlorothalonil-degrading bacteria producing transparent halos were isolated using the plate culture and chlorothalonil-selective medium. Using the morphology and 16S rDNA homology, the bacteria were then classified to genussp.,sp.,sp.,sp. andsp.sp. The RB-31and RB-38were newly discovered strains with chlorothalonil degradation ability. Two strains were determined into species level as. And their specific physiological properties were studied. Secondly, the genomic library of strainRB-38 was successfully constructed in the pUC19 vector usingcoil DH10B as the host strain, where about 10 000 clones were obtained from selective culture. A 3 494 bp of desired fragment was isolated from the library using the functional ability to degrade chlorothalonil. In the desired fragment, three open reading frames (ORFs) were tentatively identified by ORF findings and BLAST alignment on NCBI. Specifically, ORF3 encoded a hydrolytic dehalogenase. Through subcloning of this reading frame, it was proved that the degradation function of chlorothalonil was catalyzed by the enzyme encoded in this region, and no other regulation regions were required for its expression. Two ORFs upstream ofgene showed that ORF1 encoded a transposase, whereas, ORF2 encoded on IstB-like ATP-binding protein. Two ORFs were flanked by 20 bp terminal inverted repeat sequences (IR). The complete sequence presented a perfect structural similarity to IS21 transposon family members that all contain transposase coding region, ATP-binding protein coding region, and flanked by inverted repeat sequences. A new member of this family was discovered and designated as IS. Thegene was closely associated with the insertion sequence, to construct a catabolic transposon. Finally, thegene and the upstream ISfragment were cloned and identified from several genomic DNA of chlorothalonil-degrading bacteria using a PCR strategy. It infers that the sequence element of ISwas the molecular basis for the horizontal transfer in thegenes, leading that the gene exchange can occur among these degrading species. This study can enrich the chlorothalonil-degrading bacterial library, and to clone hydrolytic enzyme genes that played a key role in degrading from the genetic level. A dispersing mechanism of degrading gene was also proposed among different genus bacteria. This preliminarily clarified the functional gene and its distribution in degrading bacteria.
soils; pesticides; bacteria; gene cloning; horizontal drift
任晓洁,贺壮壮,单昕,等. 百菌清降解菌的分离鉴定及功能基因分析[J]. 农业工程学报,2020,36(19):209-216. doi:10.11975/j.issn.1002-6819.2020.19.024 http://www.tcsae.org
Ren Xiaojie, He Zhuangzhuang, Shan Xin, et al. Isolation, identification and functional gene analysis of chlorothalonil degrading bacteria[J]. Transactions of the Chinese Society of Agricultural Engineering (Transactions of the CSAE), 2020, 36(19): 209-216. (in Chinese with English abstract) doi:10.11975/j.issn.1002-6819.2020.19.024 http://www.tcsae.org
10.11975/j.issn.1002-6819.2020.19.024
S482.2
A
1002-6819(2020)-19-0209-08
2020-05-30
2020-09-13
重庆市技术创新与应用发展重点项目(cstc2019jscx-gksbX0113);国家博士后基金面上项目(2019M662362);山东省自然科学基金博士项目(ZR2019BC099);国家博士后基金面上项目(2019M650167);山东省博士后创新项目(201902048)
任晓洁,博士,讲师,主要从事发酵工程方面的研究。Email:renxiaojie2020@163.com
赵新河,博士,讲师,主要从事发酵工程方面的研究。Email:zhaoxinhe@sdut.edu.cn
猜你喜欢
河南医学研究(2022年19期)2022-10-19
分子诊断与治疗杂志(2022年4期)2022-05-30
农药学学报(2022年2期)2022-04-09
草地学报(2022年3期)2022-03-28
安徽农业科学(2022年3期)2022-03-04
今日农业(2021年11期)2021-11-27
国际消化病杂志(2021年1期)2021-03-05
中国食用菌(2020年11期)2021-01-18
股市动态分析(2018年48期)2018-07-16
食品工业科技(2014年23期)2014-03-11
