
乙型肝炎病毒核心蛋白变构调节剂的研究进展
2020-12-15 06:28陈诗琦毛日成张继明
微生物与感染 2020年5期
陈诗琦,毛日成,张继明
复旦大学附属华山医院感染科,上海 200040
乙型肝炎病毒(hepatitis B virus, HBV)是一种小型DNA病毒。全世界约有2.4亿慢性HBV感染者,每年因各种严重的肝病〔包括肝硬化、肝细胞癌(hepatocellular carcinoma, HCC)和肝功能衰竭等〕导致的死亡数达 686 000 人[1-2]。目前仅有两类药物被批准用于慢性乙型肝炎(chronic hepatitis B, CHB)的治疗,即核苷(酸)类似物(NAs)和聚乙二醇干扰素-α(PEG-IFN-α)。前一类药物通过抑制HBV聚合酶的活性,干扰前基因组RNA(pgRNA)到病毒DNA的反转录过程。NAs可以抑制HBV DNA的合成,但对于消除共价闭合环状DNA (covalently closed circular DNA,cccDNA)和钝化免疫反应无效。后一类药物PEG-IFN-α虽然影响病毒复制的多个步骤,但其疗效适中[3],且药物耐受性较差。目前CHB功能性治愈的标准是:血清乙型肝炎表面抗原(hepatitis B surface antigen, HBsAg)和HBV DNA持续检测不到,乙型肝炎e抗原(hepatitis B e antigen,HBeAg)转阴,伴或者不伴HBsAg血清学转换,肝脏炎症缓解和组织病理学改善[4]。然而长期NAs治疗虽可以降低HBsAg水平,但HBsAg阴转率仅为0~3%,PEG-IFNα 单药治疗的HBsAg阴转率稍高,约为3%~7%。因此,需要新的治疗方法来达到CHB的功能性治愈。
HBV核心蛋白是HBV核衣壳的组成部分,具有重要的生物学特性和功能,且在T细胞和B细胞水平上,HBV核心蛋白的抗原性较HBeAg的更为重要。因此HBV核心蛋白已逐渐成为抗HBV治疗的新的干预目标。本文针对HBV核心蛋白的结构、功能,结合HBV核心蛋白变构调节剂的类型和功能等方面,对近年来靶向HBV核心蛋白治疗CHB的研究现状进行综述。
1 HBV核心蛋白的结构
HBV核心蛋白由183个氨基酸残基构成,分子量约为 21 000。在HBV复制期间,mRNA从HBVC基因开始转录,该基因位于HBV的开放阅读框上。翻译从C基因的第2个密码子AUG开始,起始149个氨基酸残基形成富含α螺旋的装配结构域,也称为球状N末端结构域(N-terminal domain,NTD);而末端的34个氨基酸残基形成精氨酸C末端结构域[5](C-terminal domain,CTD)。HBV核心蛋白单体经折叠和稳定化后,形成具有四螺旋束的同型二聚体[3,6],最终组装为20面体的核衣壳。
2 HBV核心蛋白的功能
2.1 核衣壳的组装
HBV核心蛋白最主要的功能是组装HBV核衣壳。每个二聚体通过弱疏水作用与4个相邻亚基结合[7],最终组装成的核衣壳为20面体。组装成的核衣壳分为两种类型:多数是由240个HBV核心蛋白(即120个同型二聚体)组装而成;少数核衣壳的尺寸较小,含有180个HBV核心蛋白(即90个同型二聚体)。在乙型肝炎患者体内,这两种颗粒均存在,其中体积稍大的颗粒占多数。NTD对于同型二聚体之间的组装连接是必需的,而即使从HBV核心蛋白中删除CTD,核衣壳组装也不会受到影响[8]。虽然核衣壳组装不需要CTD,但后者仍然涉及许多其他功能。
2.2 pgRNA的包装和逆转录
由超螺旋的cccDNA分子转录的pgRNA离开宿主细胞核并在细胞质中结合反转录酶,最后包装到核衣壳中,即衣壳化。核衣壳内HBV核心蛋白的CTD对于pgRNA衣壳化是必需的。体外研究表明,缺乏CTD的HBV核心蛋白不会包裹pgRNA[9]。pgRNA反转录为rcDNA包含多个步骤:模板转换、引物易位和DNA延伸。这些步骤均发生于衣壳内。HBV核心蛋白上的CTD参与rcDNA的合成[10],当这种富含精氨酸的结构域CTD发生突变时,rcDNA不能合成。
2.3 病毒颗粒的形成和分泌
成熟的核衣壳与定位于内质网后、高尔基复合体前腔的HBsAg结合后,形成完整的病毒颗粒从核糖体释放出来[11]。HBV核心蛋白负责选择成熟的核衣壳,即含有rcDNA包膜的核衣壳。
2.4 cccDNA的扩增和调节
rcDNA也可转移回宿主细胞核进行再循环,以补充cccDNA库。从核衣壳释放的rcDNA进入宿主细胞核,在DNA聚合酶的作用下,转化为cccDNA。除了帮助rcDNA向宿主细胞核的转运外,核衣壳在调节cccDNA中起着额外的作用。HBV核心蛋白也是cccDNA微染色体的一个组成部分[12],可通过表观遗传机制对cccDNA的活性进行调节[13]。HBV核心蛋白可以改变cccDNA-组蛋白复合物的核小体间距,已有体内和体外研究表明,这与核小体数量的变化相关[12,14]。另一种调节机制是HBV核心蛋白可诱导cccDNA中CpG岛2的低甲基化,该区域甲基化如增强将导致cccDNA活性降低[15]。染色质免疫沉淀(chromatin immunoprecipitation assay, ChIP试验)显示,HBV核心蛋白优先与该区域结合。当HBV核心蛋白与cccDNA中的CpG岛2结合时,它是低甲基化的,从而导致cccDNA的活性增强,转录增加[13]。
2.5 与宿主免疫反应的相互作用
除了通过上述机制增强cccDNA转录外,HBV核心蛋白还参与IFN-α介导的cccDNA的破坏[16]。IFN-α可以激活一种核胞苷脱氨酶APOBEC3A,APOBEC3A与cccDNA结合的HBV核心蛋白直接相互作用,可使cccDNA易于被核酸酶降解。此外,HBV核心蛋白与干扰素刺激基因(interferon-stimulated genes,ISGs)之间的相互作用也参与其中。在一些细胞系研究中,已发现HBV核心蛋白抑制双链DNA介导的IFN效应[17]。这种抑制的先决条件是HBV核心蛋白的核定位可抑制ISGs的转录[18]。HBV核心蛋白还与肿瘤坏死因子相关凋亡诱导配体(TNF-related apoptosis-inducing ligand,TRAIL)相互作用,其结果与HBV感染的肝细胞死亡有关。过表达的HBV核心蛋白还可降低TRAIL诱导的人肝癌细胞凋亡[19]。HBV核心蛋白同时也是B细胞、T辅助细胞和细胞毒性T淋巴细胞(cytotoxic lymphocyte,CTL)免疫介导的病毒消除的靶点[20]。CTL和T细胞表位在NTD和CTD上均可表达,而B细胞表位主要在NTD上表达。研究已显示这些表位中的突变会影响宿主免疫应答[21, 22]。CTD的CTL表位磷酸化位点的突变与肝纤维化相关[23]。HBV核心蛋白通过与宿主免疫因子相互作用对病毒活性和肝细胞破坏发挥复杂的调节作用。
3 核心蛋白变构调节剂(CpAMs)
核心蛋白涉及HBV复制的多个步骤,是高度保守的病毒蛋白,由于它没有同源物,因此是具有病毒特异性的良好治疗靶标。CpAMs是通过变构机制干扰核衣壳组装的小分子,可以有效地降低病毒载量和病毒抗原。从作用机制上进行分类,CpAMs可以分为两类:Ⅰ类CpAMs,包括氨磺酰苯甲酰胺(SBAs)、苯丙烯酰胺[24](PPAs)、吡唑基-噻唑(PT)、乙二酰胺-吡咯烷(GPA)和二苯并-硫氮杂-2-酮(DPT)等,可诱导形态完整空核衣壳的组装;Ⅱ类CpAMs,包括杂芳基嘧啶[24](HAPs),可引起异常核衣壳的形成。在迄今被筛选分类的CpAMs中,只有HAPs属于Ⅱ类CpAMs。
3.1 甲磺酸莫非赛定(GLS4)
GLS4是由我国广东东阳光药业有限公司自主研发的新一代二氢嘧啶类药物,其作用机制是干扰HBV核衣壳的组装,可呈剂量依赖性地减少病毒核衣壳的正确组装,加速异常核衣壳形成,使病毒核衣壳和核心蛋白二聚体的量均减少,从而抑制HBV的复制及成熟病毒颗粒的产生。
研究表明,GLS4不仅可以高效阻断核衣壳的组装过程,对于已组装完成的核衣壳同样具有破坏作用,且可对包含双链DNA的成熟核衣壳特异性地进行降解[25]。冷冻电镜观察提示,GLS4对核衣壳的降解无须宿主细胞蛋白降解系统的参与[25]。在基于HepG2-NTCP细胞的HBV感染模型中,GLS4预处理可高效阻断HBV感染后cccDNA的从头合成及后续的病毒抗原表达,而对照药物恩替卡韦则无此活性。
GLS4的临床前体内、外实验结果均表明,其抗病毒效果明显优于拉米夫定,且对阿德福韦酯耐药株(rtA181T/V、rtN236T)、拉米夫定耐药株(rtM204I、rtM204I+V173L)、替比夫定耐药株(rtM204I、rtL180M+M204V)、恩替卡韦耐药株(rtM204I+S202G、rtM204I+S202G+M250V)均有明显抑制作用[26]。
GLS4目前已完成单剂量递增、连续给药剂量递增、食物对药代动力学的影响3项Ⅰ期临床试验。安全性和药代动力学研究数据表明,GLS4在很大的给药剂量范围内均有良好的安全性和耐受性,口服GLS4可达到有效药物浓度。Ⅰ期临床试验观察到的不良事件种类、发生率和严重程度在GLS4组和安慰剂组相似,且所有不良事件均为轻度,未经处理后均恢复正常或改善[26]。没有受试者因不良事件而提前终止试验。
GLS4的Ⅱa期临床试验表明其对于HBeAg阳性和阴性患者的HBV DNA复制均有明显的抑制作用,能显著降低血清HBcAg和HBV RNA水平[27]。GLS4在CHB患者中耐受良好,大部分不良事件均较轻,且无剂量限制毒性。
3.2 ABI-H类药物
ABI-H类药物是美国Assembly公司在研的新型CpAMs,包括一代药物ABI-H0731和二代药物ABI-H2158。
2017年亚太肝脏研究学会(The Asian Pacific Association for the Study of the Liver, APASL)上首次公布了ABI-H0731的Ⅰa期临床研究数据,Ⅰa期临床研究主要评估了ABI-H0731在健康志愿者中的安全性、耐受性和药代动力学[28]。随后,2018年美国肝病学会(American Association for the Study of Liver Diseases, AASLD)的年会上报告了该药Ⅰb期临床试验的最终结果,研究主要评估该药在CHB患者中的安全性、药代动力学和药效学[29]。总体而言,ABI-H0731在健康志愿者和CHB患者中的耐受性良好,没有严重的不良反应,临床没有显著的药物相关依赖。因此,研究人员认为,ABI-H0731是一种安全且耐受良好的CpAMs,具有剂量依赖性抗病毒作用,剂量范围为每日 100~400 mg。
在2019年欧洲肝病学会(The European Association for the Study of the Liver, EASL)年会上,研究者报告了ABI-H0731的Ⅱa期临床试验结果[30]。在Ⅱa期临床试验中,将受试者分为2组进行研究,201组研究招募了47例HBeAg阳性患者和26例阴性患者,已提前使用NAs治疗;202组研究招募了25例HBeAg阳性初治患者,以恩替卡韦+ABI-H0731∶恩替卡韦+安慰剂=1∶1比例随机分配。研究过程中,受试者耐受状态良好,不良事件一般为轻度或中度。201组研究中,第12周时ABI-H0731+NAs组HBV RNA水平下降2.6log10IU/ml,安慰剂+NAs组HBV RNA水平下降0.2log10IU/ml。202组研究中,第12周时恩替卡韦+ ABI-H0731组HBV DNA/RNA水平降低了4.5/2.6log10IU/ml,恩替卡韦+安慰剂组HBV DNA/RNA水平降低3.2/0.4log10IU/ml。受试者血清HBeAg和HBsAg水平均有所下降,但在早期阶段尚且不能得出有意义的结论,需后续延长用药时间进一步观察。ABI-H0731+NAs治疗耐受性良好,并在治疗的早期即显示出了抗病毒疗效以及潜在的抗病毒益处,包括HBV RNA的减少——这是单独使用NAs治疗时未见到的。
ABI-H2158是Assembly公司在研的二代CpAMs。在2019年EASL上,研究者首次报道了ABI-H2158的Ⅰa期临床试验结果,证实其耐受性良好,治疗期间出现的不良反应均为轻度[31]。药代动力学结果表明ABI-H2158口服给药可被快速吸收,消除半衰期约为10~18小时。预计其将进入 Ⅰb 期临床研究。
3.3 AB类药物
AB类药物是加拿大Arbutus公司在研的CpAMs,包括一代药物AB-423和二代药物AB-506。
2016年的EASL上首次报告了针对AB-423的研究,该研究认为AB-423是一种很有前途的药物,在细胞层面上可以有效抑制HBV的复制。在HBV感染小鼠模型中联合使用恩替卡韦具有有效抑制HBV复制的作用[32]。
2016年AASLD上报告[33, 34]了AB-423与其他抗病毒药物的联合抗病毒疗效。研究者发现,在HepAD38细胞系中, AB-423和第2代siRNA药物ARB-1740联合使用具有协同抗rcDNA活性[35]。在高压尾静脉注射小鼠模型和慢性感染人源化肝细胞小鼠模型中,这种双靶点作用的联合用药使得其抑制HBV复制的能力更强。
AB-506是Arbutus公司的在研的二代CpAMs。 2018年EASL上报告了AB-506与NAs或ARB-1467联合应用的体外双重抗病毒效果[36]。在高压尾静脉注射小鼠模型中,AB-506+AB-452(HBV RNA去稳定剂),AB-506+TDF和AB-452+TDF的双重组合表现出强烈的抗病毒活性,其下调血清HBV DNA的平均值分别为1.4log10IU/ml、1.9log10IU/ml和2.2log10IU/ml。同时,3组药物联合使用,即AB-506+AB-452+TDF,降低血清HBV DNA的能力更强,血清HBV DNA下降约为2.8log10IU/ml。所有AB-506和AB-452治疗组均显示肝脏HBV DNA减少,TDF单药组观察到的肝脏HBV DNA减少程度相对较少。AB-506与NAs或ARB-1467的体外双重联合用药在降低HBV rcDNA和HBsAg水平上也有累积至中度协同作用,但对细胞活力则没有显著影响。
以上临床研究数据表明,这些药物在联合使用时具有协同的体外和体内抗病毒活性,并且可能用于将来的联合治疗方案中。
3.4 RO7049389(RG7907)
RO7049389是一种Ⅱ类小分子CpAMs,它可通过诱导异常HBV核心聚集体的形成,导致有缺陷的衣壳组装,从而抑制HBV复制。
在2018年EASL上,研究者报告了RO7049389的单递增剂量(single ascending dose,SAD)组(5个剂量组,150~2 000 mg)和多递增剂量(multiple ascending dose,MAD)组(5个剂量组, 200~800 mg,每日2次)在健康志愿者中的安全性和药代动力学研究的结果[37]。在整个给药剂量范围内,RO7049389可以被迅速吸收并从血浆清除,极少产生累积。健康志愿者对于RO7049389的耐受性良好,极少有不良事件发生。此外,对6名CHB患者给予药物200 mg, 每日2次,口服28 d,可观察到受试者HBV DNA从给药前水平开始稳定地持续下降,中位数(最大)下降为2.7(3.4)log10IU/ml,3/6例患者的HBV DNA水平低于检测下限。在整个治疗过程中,没有观察到病毒学反弹。
在2019年EASL上,研究者报告了RO7049389针对CHB患者在不同给药剂量条件下的抗病毒效果[38],3组队列分别为:200 mg、400 mg和600 mg,每日给药2次,给药时间28 d。所有队列均显示出HBV DNA和HBV RNA显著下降,给药期间HBsAg或HBeAg水平没有显著变化,CHB患者对于RO7049389耐受性良好。预计下一步会将该药与其他抗病毒药物联合使用,并进一步评估联合用药的抗病毒效果。
3.5 JNJ类药物
美国Janssen公司在研的CpAMs主要为JNJ-64530440(JNJ-0440)和JNJ-56136379(JNJ-6379)。
在2018年AASLD上,研究人员首次报道了JNJ-0440的体外抗病毒活性和作用方式[39,40]。JNJ-0440通过诱导形成空的但形态完整的病毒核衣壳来抑制HBV的复制。体外实验证实JNJ-0440以剂量依赖性的方式阻断PHHs中cccDNA的从头合成,降低细胞培养上清液中HBeAg和HBsAg的水平。此外JNJ-0440的药物安全性、耐受性和药代动力学实验均已在Ⅰ期临床研究中完成评估(NCT03439488),没有因发生不良事件而终止研究。在2019年EASL上公布的Ⅰ期临床研究数据显示,JNJ-0440的耐受性和安全性良好。目前该药正在CHB患者中进行疗效评估[41]。
JNJ-6379首项人体临床研究(NCT02662712)的Ⅰa期研究结果提示该药在健康受试者中安全性和耐受性良好。在2018年AASLD上,研究者公布了该项研究的Ⅰb期临床研究数据,即多剂量JNJ-6379 用于CHB患者治疗的安全性、药代动力学和抗病毒活性研究[42]。研究纳入了初治、无肝硬化的HBeAg阳性或阴性CHB患者,随机分配至JNJ-6379治疗组或安慰剂治疗组28 d,随访8周。4种JNJ-6379治疗组的用药剂量分别为每日25 mg、75 mg、150 mg、250 mg。整个治疗周期中,JNJ-6379安全性和耐受性良好,所有剂量组均表现出较强的抗HBV活性。
在2018年全球肝炎峰会(Global Hepatitis Summit 2018)上公布的部分Ⅱa期临床研究结果显示,使用JNJ-6379后的第29天,在所有治疗组中均观察到HBV DNA和HBV RNA从基线显著下降,但没有观察到HBsAg的显著变化,故需延长用药时间来进一步观察。在2019年APASL上公布的结果提示JNJ-6379耐受性良好,在整个剂量范围内(25 mg、75 mg、150 mg、250 mg)产生相似的药代动力学[43]。在所有剂量中,每周接受75mg剂量在亚洲和非亚洲的CHB患者中抗病毒效果相当。这些数据将支持其在欧洲和亚洲的更大队列中进一步评估相同剂量的JNJ-6379±NAs的抗病毒效果(NCT03361956)。
3.6 其他CpAMs
除上述CpAMs外,还有包括中国齐鲁药业研发的QL-0A6a。细胞实验的研究结果提示,低浓度的QL-0A6a可干扰HBV核衣壳的装配[44]。QL-0A6a 对不同基因型的HBV均有抑制作用,与NAs联合使用时,具有协同作用。QL-0A6a对NAs耐药的突变株也具有抗病毒效果。QL-007也是齐鲁药业在研的CpAMs,该药目前已进入Ⅰ期临床研究。
GLP-26是美国Emory University在研的CpAMs,2018年EASL上关于GLP-26的临床前研究显示其具有良好的抗病毒效果,与现有抗病毒药物的联合用药具有协同作用。GLP-26目前仍处于临床前研究阶段[45]。
EP-027367是美国Enanta公司在研的CpAMs。2019年EASL公布的研究结果显示,EP-027367 仅需亚微摩尔的剂量即可抑制cccDNA的从头合成。该药目前仍处于临床前研究阶段。
4 CpAMs的应用和展望
目前临床应用的抗HBV药物很少能达到功能性治愈。新的治疗方案,应该针对病毒生命周期的多个步骤、多个靶点,且应该在无毒浓度下抑制HBV复制。核心蛋白变构调节剂作为新型的抗病毒药物,其抑制HBV作用显著,耐受性好,安全性高,可作为新的乙型肝炎治疗用药。此外CpAMs可能使得预先形成的衣壳不稳定,因此可用于血液制品以减少HBV传播的风险[46]。表1总结了目前正在研发的CpAMs关键信息。
尽管针对慢性HBV感染有各种可供选择的治疗手段,但没有一种是完全令人满意的。目前有5种被批准用于治疗慢性HBV感染的NAs:拉米夫定、替比夫定、恩替卡韦、阿德福韦和替诺福韦[47]。它们抑制HBV的反转录,可以将血液中病毒含量降低至不可检测的水平。然而,NAs单一疗法很少能够根除病毒[48];且由于频发的耐药性问题,NAs的临床应用也受到限制。而PEG-IFNα虽然具有无耐药性、免疫刺激和直接抗病毒等特性,但仅在1/3的患者中有效[4]。由于CHB的难治愈性问题,近些年国内外均对CHB的治愈概念进行了不少探讨以期逐步实现彻底治愈的目标。在近些年的CHB临床治愈的专家共识中,也对HBsAg的阴转甚至出现抗-HBs阳转提出了要求[4],这是现有的临床治疗药物所无法达到的标准。目前在研药物的临床试验检测过程中,多将HBsAg作为监测效用的生物标志物之一,代表药物有RNAi、CpAMs等。
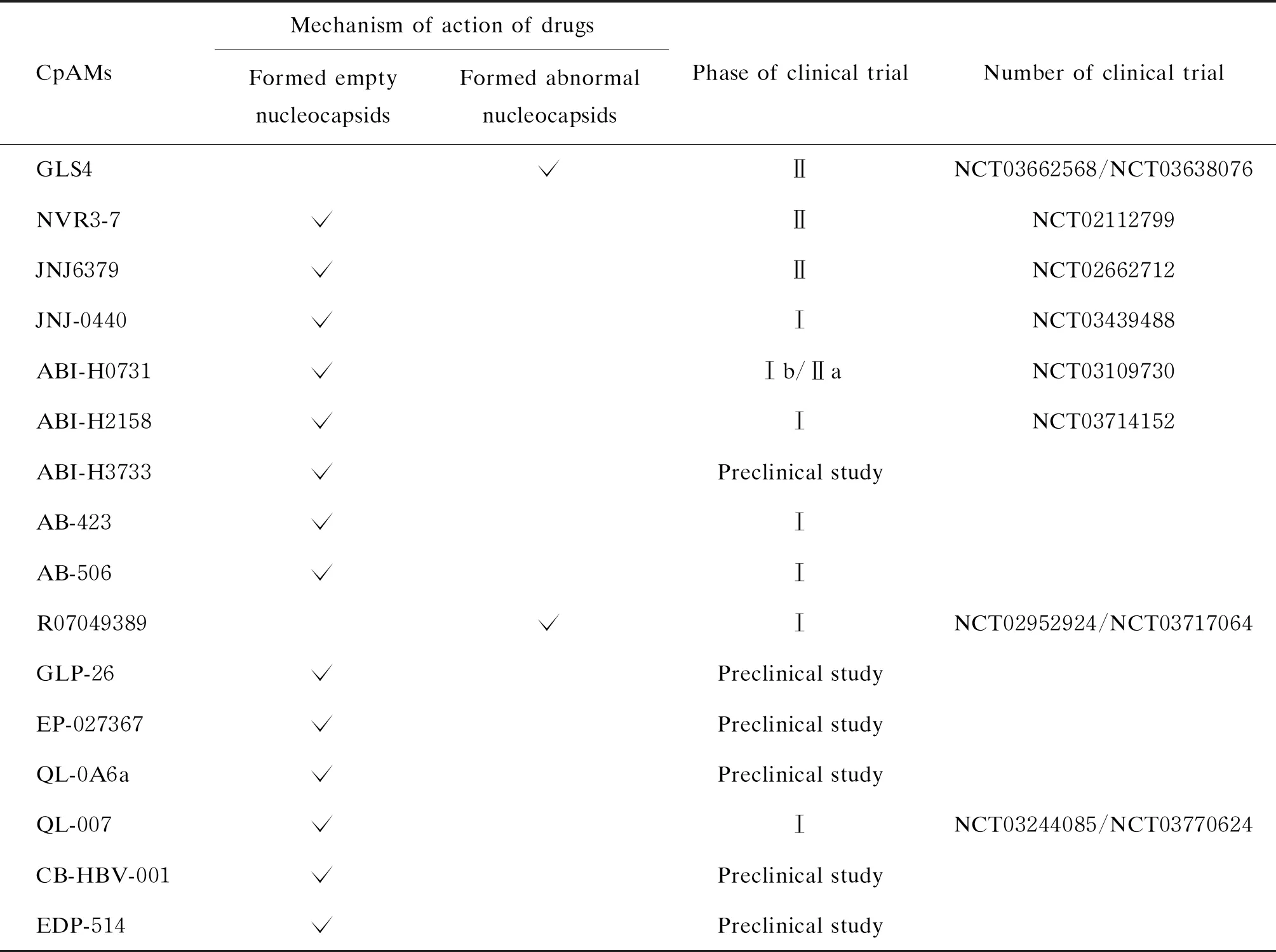
表1 临床试验阶段中的CpAMs
CpAMs代表着一种新型的抗病毒药物,与以往的抗病毒药物形成鲜明的对比。如目前在对于CpAMs的研究中,暂时未发现药物的毒性效应;在药物治疗期间,也未出现病毒学反弹;对于NAs产生耐药性的细胞株,CpAMs也能产生抗病毒效应。
CpAMs作为一种新型抗病毒药物,因其作用靶点不同于NAs和PEG-IFN-α,故联合应用时具有协同作用,抗病毒活性增强,抗-HBs阳转,患者在随访期间亦未出现病毒学反弹,有望达到功能性治愈,这为CHB的治疗提供了新的治疗方向,有望造福更多的CHB患者。
猜你喜欢
全科护理(2022年10期)2022-12-26
中国合理用药探索(2022年1期)2022-11-26
肝博士(2022年3期)2022-06-30
今日农业(2021年21期)2021-11-26
中老年保健(2021年12期)2021-08-24
乡村科技(2021年33期)2021-03-16
国际放射医学核医学杂志(2021年10期)2021-02-28
四川蚕业(2020年4期)2020-02-10
现代农业科技(2016年20期)2016-12-20
现代养生·下半月(2015年6期)2015-09-07
