
湖南益阳黑茶中桔青霉线粒体全基因组序列测定及分析
2020-12-15 05:51:24胡治远刘素纯徐正刚刘石泉文欣
茶叶科学 2020年6期
胡治远,刘素纯,徐正刚,刘石泉,文欣
湖南益阳黑茶中桔青霉线粒体全基因组序列测定及分析
胡治远1,2,刘素纯1*,徐正刚2,刘石泉2,文欣3
1. 湖南农业大学食品科学技术学院,湖南 长沙 410128;2. 湖南城市学院材料与化学工程学院,湖南 益阳 413000;3. 湖南省食品质量监督检验研究院,湖南 长沙 410111
对黑茶中分离的一株桔青霉线粒体基因序列进行测定与分析,并探究其与近缘种微生物的系统发育关系。结果表明,桔青霉线粒体基因组是一条全长27 537 bp的环状DNA分子,共编码42个基因(15个蛋白质编码基因,2个rRNA,24个tRNA以及1个独立的ORF),基因组碱基构成为:A(36.17%)、T(37.06%)、C(11.82%)、G(14.95%)。15个蛋白质编码基因均采用典型的ATG作为起始密码子、TAA或TAG作为终止密码子,基因排列顺序与已报道的青霉属物种相似,在进化上较为保守。蛋白质编码基因编码频率较高的氨基酸分别为Leu、Ile、Ser和Phe;RSCU频率最高的4个密码子依次是UUA、AUA、UUU和GGU。24个tRNA基因存在30处G-U错配,均可形成典型三叶草结构。系统发育分析结果表明,桔青霉分类地位上关系最密切是ShG4C,其次是与。
黑茶;桔青霉;线粒体基因组;系统发育
线粒体(Mitochondrion)是存在于真核生物细胞中的一种半自主性细胞器,其基因组独立复制,不受减数分裂时核染色体重组的影响。线粒体基因组具有母系遗传、进化速率较快等特点,其基因组成、遗传密码和复制方式,记载着生物演化过程中丰富的进化信息,是研究物种起源与进化的重要材料[1]。目前,线粒体基因组信息分析技术已成为一种重要的分子标记手段,被广泛应用于分类鉴定、群体遗传关系、种间分子进化等的研究中[2],作为传统分类法的补充。
桔青霉()是青霉属的一种丝状真菌,其分布较为广泛,在水果、粮食及蔬菜等基质中均可正常生长[3],在黑茶中则尤为常见[4-6]。在黑茶渥堆时,桔青霉生长所产生的胞外酶可促进茶叶内含成分转化[7],对推动发酵有一定的积极作用。但在其他加工阶段或是成品茶叶中,桔青霉大量繁殖则会导致品质劣变[8],其一定条件下甚至可分泌对肾脏及免疫系统具有毒性的桔霉素[9-10],一般被认为是黑茶中的污染性微生物。近年来,黑茶及其深加工产业快速崛起,其产销量已跃居我国六大茶类的第二位,由微生物污染所导致的产品真菌毒素超标等安全隐患正受到业界和消费者的关注。黑茶作为一种后发酵茶,对其中微生物种群的准确鉴定将是保障产品安全、提升发酵质量的重要途径之一。鉴于此,本文以湖南黑茶中分离出的一株桔青霉作为研究对象,首次对其线粒体基因组全序列进行测定与分析,并与已报道的近缘种微生物线粒体基因组信息进行对比,旨在促进这一真菌的群体遗传学、进化和分类学提供基础数据。
1 材料与方法
1.1 菌种筛分与观察
桔青霉菌株由湖南益阳所产黑茶内分离得到,茶叶样品由湖南益阳茶厂有限公司提供(品种:金湘益800 g茯砖茶,生产日期:2015年11月8日)。桔青霉菌种的筛选采用梯度稀释分离法[11]:取粉碎的茶样放入添加玻璃珠的无菌生理盐水三角瓶中,120 r·min-1振摇20 min使其中微生物散布均匀,梯度稀释后,取10-4~10-5稀释液涂布于PDA平板,28℃培养4 d,从长势较佳的桔青霉菌落挑取菌体于PDA平板上划线以获得纯培养的菌落,观察其菌落形态与光学显微镜下特征。此外,挑取生长状况适宜的菌体制成固定标本[11],采用离子溅射仪对样品表层进行喷金处理后,在扫描电镜(Zeiss Sigma 300)下观察其结构。
1.2 线粒体DNA的提取
将桔青霉接种至不添加琼脂的马铃薯蔗糖液体培养基,置28℃环境下振荡培养4 d后,离心去除培养液,获得适量菌体作为提取DNA的材料。桔青霉线粒体基因组采用DNeasy Plant Mini Kit(Qiagen,Valencia,CA)提取,获取DNA后,采用双链DNA超敏试剂盒(Invitrogen,Qubit dsDNA BR检测试剂盒,Q32853)中的Qubit荧光计检测总DNA质量[12]。
1.3 线粒体DNA序列测定
样品的测序工作由深圳市惠通生物科技有限公司完成,基于Illumina miseq 2500平台(Illumina,San Diego,CA)进行,所有片段均为双向测序,未能测通的序列则采用克隆测序或重新设计引物再测序,补全序列适配器和低质量的reads使用NGS QC工具包删除[13]。共获取了8.3 G原始测序数据,移除连接物和未配对、短小、质量差的reads后,将高质量的reads用IDBA-UD进行从头组装[14]。
1.4 基因组注释与tRNA预测
使用在线软件MITOS web server(http://mitos.bioinf.uni-leipzig.de/index.py)对基因组进行注释[15],获得桔青霉线粒体基因组注释信息,并将结果提交至GenBank数据库中。使用在线软件OGDraw(https://chlorobox.mpimp-golm.mpg.de/OGDraw.html)对注释结果进行可视化处理[16],绘制桔青霉线粒体全基因组物理图谱。采用在线软件tRNAscan-SE 2.0(http://lowelab.ucsc.edu /tRNAscan-SE)预测tRNA的二级结构[17],无法直接预测的基因通过人工校正完成。
1.5 基因组碱基组成分析
使用在线软件Feature Extract 1.2L server(http://www.cbs.dtu.dk/services/FeatureExtract)提取桔青霉线粒体基因组的蛋白编码区、tRNA与rRNA序列[18],使用Bioedit计算不同基因4种碱基比例,并根据式(1)和(2)计算碱基偏斜。

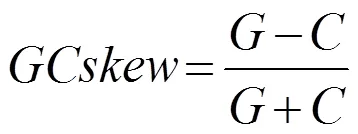
1.6 密码子使用模式分析
使用DAMBE计算蛋白质编码基因的同义密码子使用相对频率(Relative synonymous condon usage,RSCU)[19],分析密码子使用模式。
1.7 基因组微卫星序列分析
使用在线软件IMEX(http://43.227.129. 132:8008/IMEX)提取桔青霉线粒体微卫星序列[20],得到的数据根据式(3)和(4)进行相对丰度和相对密度的计算。
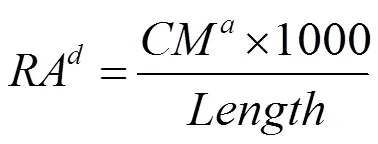
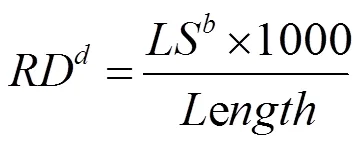
式中:为相对丰度,RD为相对密度,CM为线粒体微卫星数量,LS为线粒体微卫星序列长度。
1.8 系统发育分析
为研究桔青霉和近缘物种的关系,采用Maximum likelihood法,以属的、、;属的;属的、、、、作为外群,以及属的ShG4C、、、、、、共17种微生物线粒体蛋白质编码基因序列串联后进行系统发育分析,序列对比及系统树构建采用Mega 7.0进行[21]。
2 结果与分析
2.1 桔青霉的形态特征
桔青霉的菌落特征如图1所示,其在PDA平板上生长速度较快(图1-A),36 h即可形成绒状的小型菌落,96 h时直径达15~20 mm,菌落表面呈致密绒状,色泽为青绿色至灰绿色,边缘色泽较淡,菌落表面较平坦,中心区域有部分隆起,无渗出液产生。
在光学显微镜下(图1-B),未发现有性型结构,由菌丝异化形成的分生孢子头较为多见,呈半披张的扫帚状,具备青霉属物种的典型特征[22]。在电镜观察下,未成熟的分生孢子着生在顶囊上(图1-C),每簇分生孢子链包括3~8粒分生孢子,随时间延长而逐渐增大,孢梗茎直径约2~4 µm;成熟后的分生孢子从孢子头上脱落(图1-D),分散在基质内,孢子多呈较规则的椭球状(图1-E),外壁布满不规则突起,孢子体积较小,直径1.2~2 µm;偶有形状差异较大(图1-F)的分生孢子出现。其菌落形态、显微特征符合中国真菌志[22]中对的描述。
2.2 线粒体基因组结构及特征
桔青霉线粒体全基因组结构及特征如图2、表1所示,其是一条全长27 537 bp的双链闭合环状DNA分子(GenBank登录号:MK919205,详见Hu等[23]报道),共包含42个基因,其中有15个蛋白质编码基因(、、、、、、、、、、、、、、),2个核糖体RNA基因(、),24个转运RNA基因,以及1个独立的开放阅读框()。
桔青霉线粒体基因组的42个基因共存在2处基因重叠,一处在与之间,重叠序列为1 bp;另一处出现在与之间,其中被所隔开,两条基因有145 bp重叠。基因间隔现象则比较普遍,42个基因共存在38处间隔,其中最长的一处为和之间,长度达到842 bp,既无间隔又无重叠的区域只有2处。

注:A. 桔青霉菌落形态(96 h);B. 光镜下分生孢子头(200×);C. 未成熟分生孢子;D. 成熟分生孢子;E/F. 不同形态分生孢子
Note: A. Colonial morphology of(96 h). B. Conidial head in optical microscope (200×). C. Immature conidia. D. Ripe conidia. E/F. Different forms of conidia
图1 桔青霉菌落形态与显微特征
Fig. 1 Colonial morphology and microscopic characteristics of
2.3 线粒体基因组核苷酸组成
桔青霉线粒体基因组核苷酸组成情况如表2所示,全基因组A、T、C、G碱基的含量分别为36.17%、37.06%、11.82%、14.95%,具有显著的A+T偏向性。其中,24个tRNA基因序列全长为1 803 bp,A+T含量为60.90%;15个蛋白质编码基因全长为14 355 bp,A+T含量为73.33%;和长度分别为4 721和1 599 bp,A+T含量分别为71.81%和65.35%;开放阅读框()长度为1 050 bp,A+T含量为71.43%,不同类型基因均表现出对A+T碱基的显著偏向性,其含量比例符合青霉属微生物的一致特征[24]。除全基因组AT偏斜(–0.012 2)、蛋白质编码基因AT偏斜(–0.078 7)以及tRNA的AT偏斜(–0.045 5)为负值外,其余基因AT及GC偏斜均为正值。
青霉属8种微生物线粒体基因组核苷酸组成由表3所示,8种真菌线粒体基因组A+T含量均较高,且比例较为接近。其中,桔青霉线粒体基因组A+T含量处于偏低水平,依次为桔青霉(73.22%)<(74.44%)<(74.53%)<(74.57%)<(74.61%)<(74.66%)<ShG4C(74.84)<(75.06%),此外,所有青霉属物种的蛋白质编码基因和rRNA基因中A+T含量高低趋势与线粒体基本一致。在碱基偏斜方面,桔青霉与除和之外的6种青霉表现一致,均是AT偏斜为负值、GC偏斜为正值。在rRNA排列顺序上,桔青霉基因位于和间,被基因所隔开;则位于和之间,与青霉属其他物种相比[24],未出现显著移位,这些信息表明了桔青霉线粒体基因组在进化上的保守性。
2.4 线粒体基因组蛋白质编码基因
桔青霉线粒体基因组包含15个蛋白质编码基因,平均长度为957 bp,最短的长147 bp,最长为则达到1 977 bp,长度差异较显著。15个基因ATG作为起始密码子,均采用典型的TAA(、、、、、、、、、、、)或TAG(、、)作为终止密码子。按功能不同,可将蛋白质编码基因分为4类,其中编码ATP合成酶亚基相关的基因有3个(、、);编码细胞色素相关的基因有4个(、、、);编码氧化还原酶亚基相关的基因有7个(、、、、、、),以及一个核糖体蛋白编码基因。桔青霉线粒体蛋白质编码基因排列顺序与青霉属真菌ShG4C、、完全一致[25-26],在进化上较为保守。而蛋白质编码基因重排现象在其他青霉属真菌中则比较常见,如缺乏,缺乏,的基因被所替代等。
蛋白质编码基因编码蛋白的氨基酸种类及比例由图3所示,不同种类氨基酸的含量差距较大,其中编码最频繁的氨基酸是亮氨酸(Leu,14.55%),其次是异亮氨酸(Ile,10.56%)、丝氨酸(Ser,8.95%)和苯丙氨酸(Phe,8.44%),编码频率较低的11种氨基酸分别为精氨酸(Arg)、天冬酰胺(Asn)、天冬氨酸(Asp)、半胱氨酸(Cys)、谷氨酰胺(Gln)、谷氨酸(Glu)、组氨酸(His)、赖氨酸(Lys)、甲硫氨酸(Met)、脯氨酸(Pro)、酪氨酸(Tyr)含量均在5%以内。这一趋势与其他青霉属物种基本一致[25-26]。
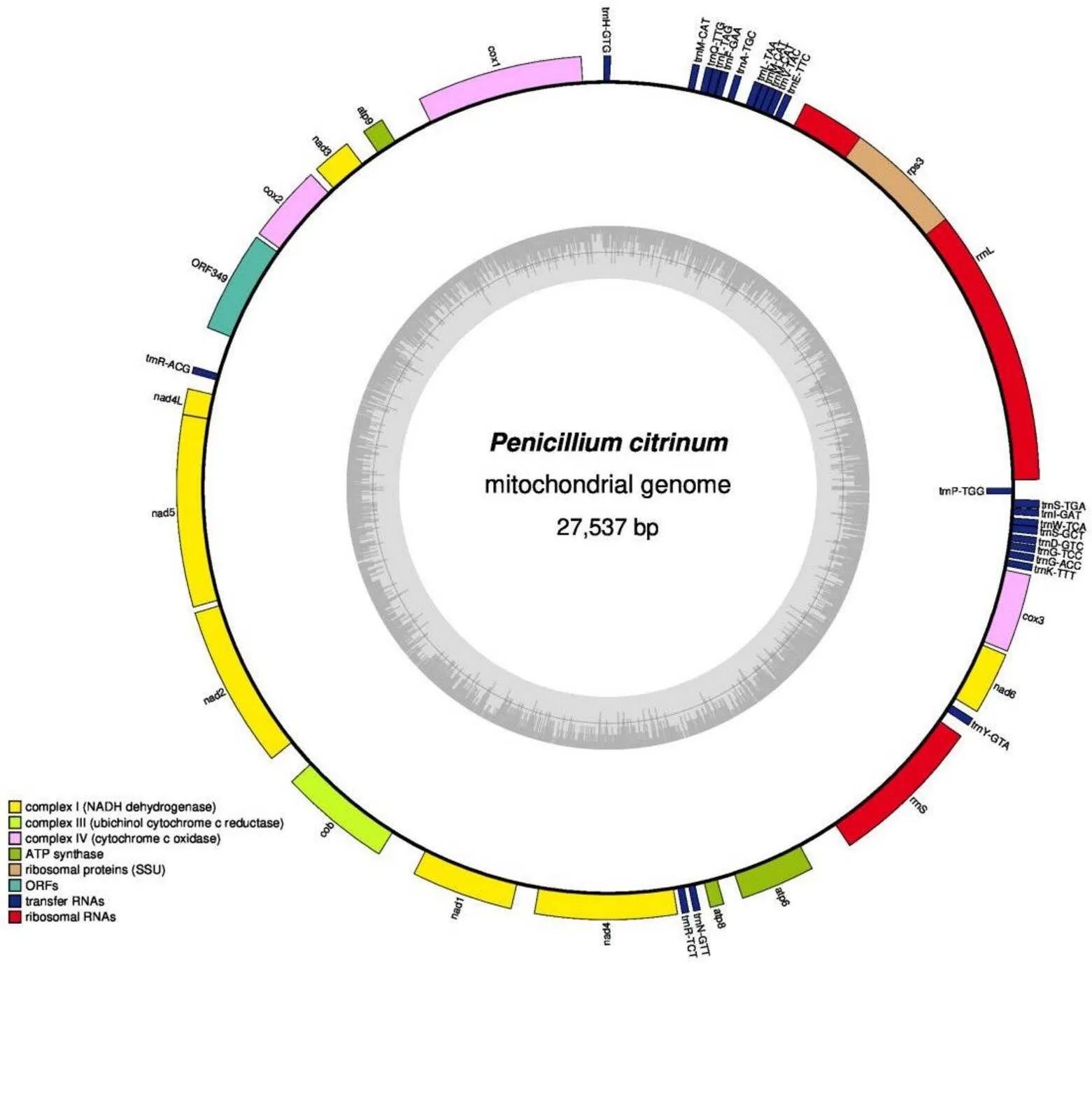
图2 桔青霉线粒体基因组结构
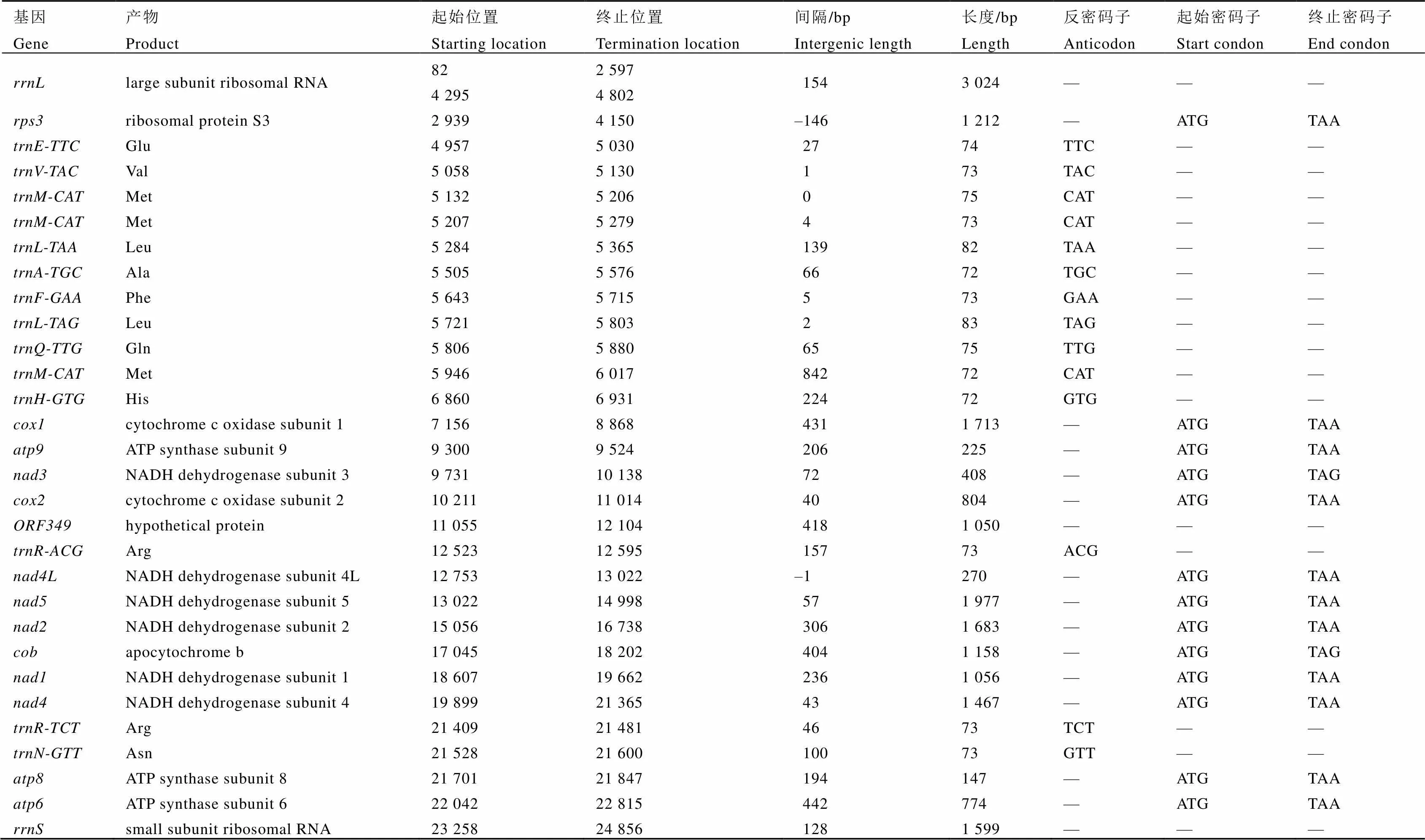
表1 桔青霉线粒体基因组成
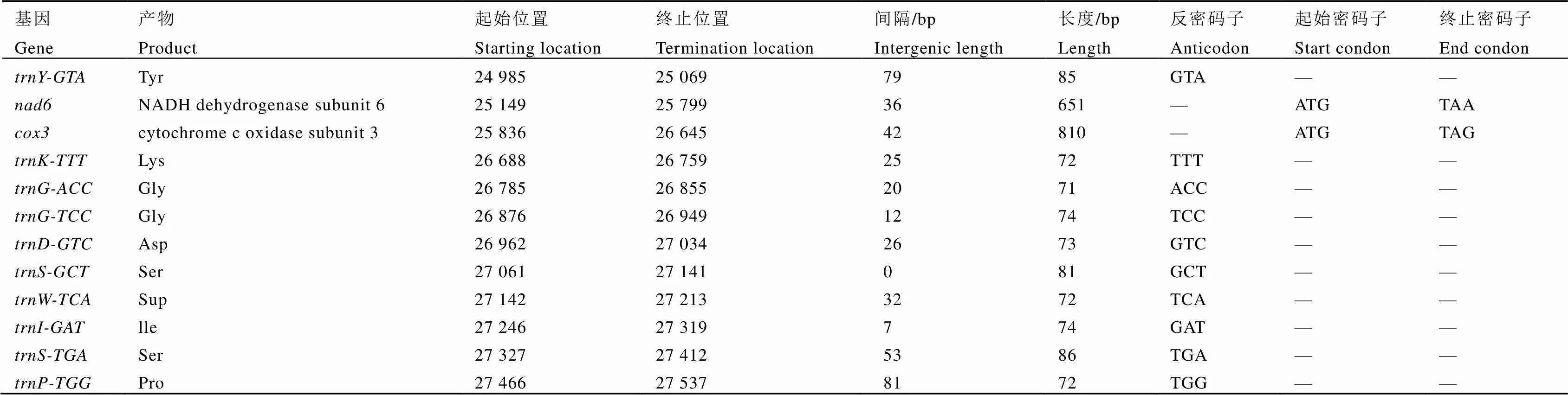
续表1

表3 青霉属8种微生物线粒体基因组核苷酸组成
桔青霉线粒体蛋白质编码基因的相对同义密码子使用频率(RSCU)如表4所示,当RSCU数值大于1时,表示密码子的使用偏好较高,小于1则反之。除去终止密码子(Stop codon),桔青霉线粒体蛋白编码区共包含4 727个密码子。密码子使用情况显示,其对A和U碱基具有显著的偏向性,其中使用次数在200次以上的4个密码子分别是UUA(Leu)、AUA(Ile)、UUU(Phe)和GGU(Gly),密码子使用次数依次为618、322、263和209,这4种密码子在青霉属微生物[27]的线粒体蛋白质编码基因中也同为高频密码子,使用次数分别为638、305、300、211[27]。而UGC、GGC、CUC、CGA、CGC、CGG、UCG、UGG 8种密码子在桔青霉线粒体蛋白质编码基因中的使用次数均为0,在相对同义密码子的使用上与[27]具有较高的相似性。
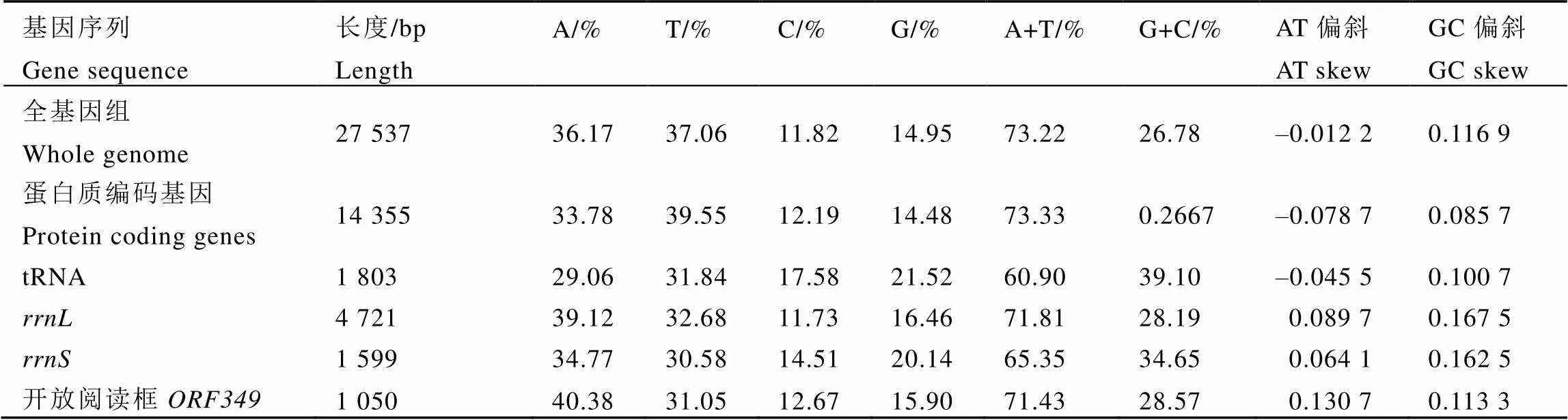
表2 桔青霉线粒体基因组核苷酸组成
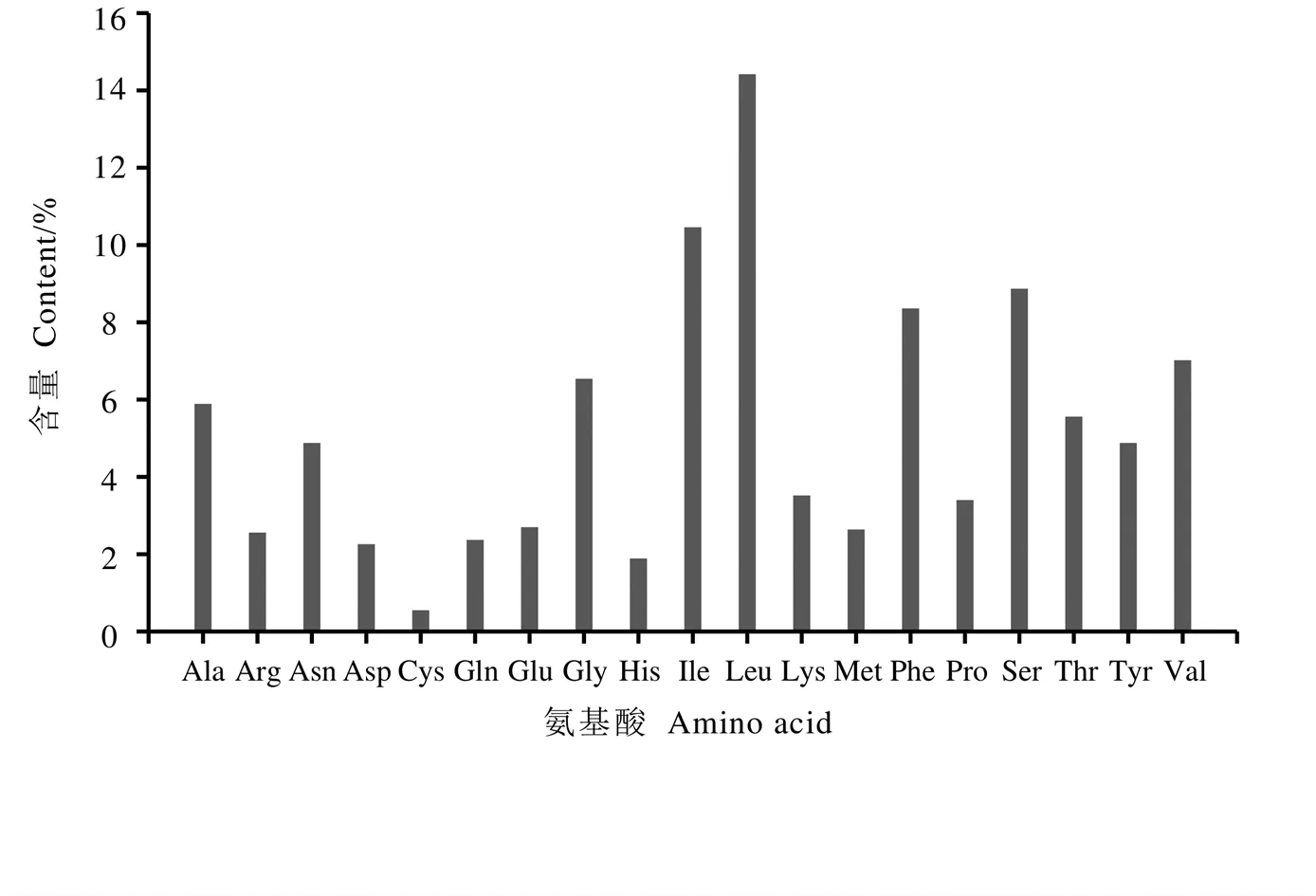
图3 桔青霉线粒体基因组氨基酸含量
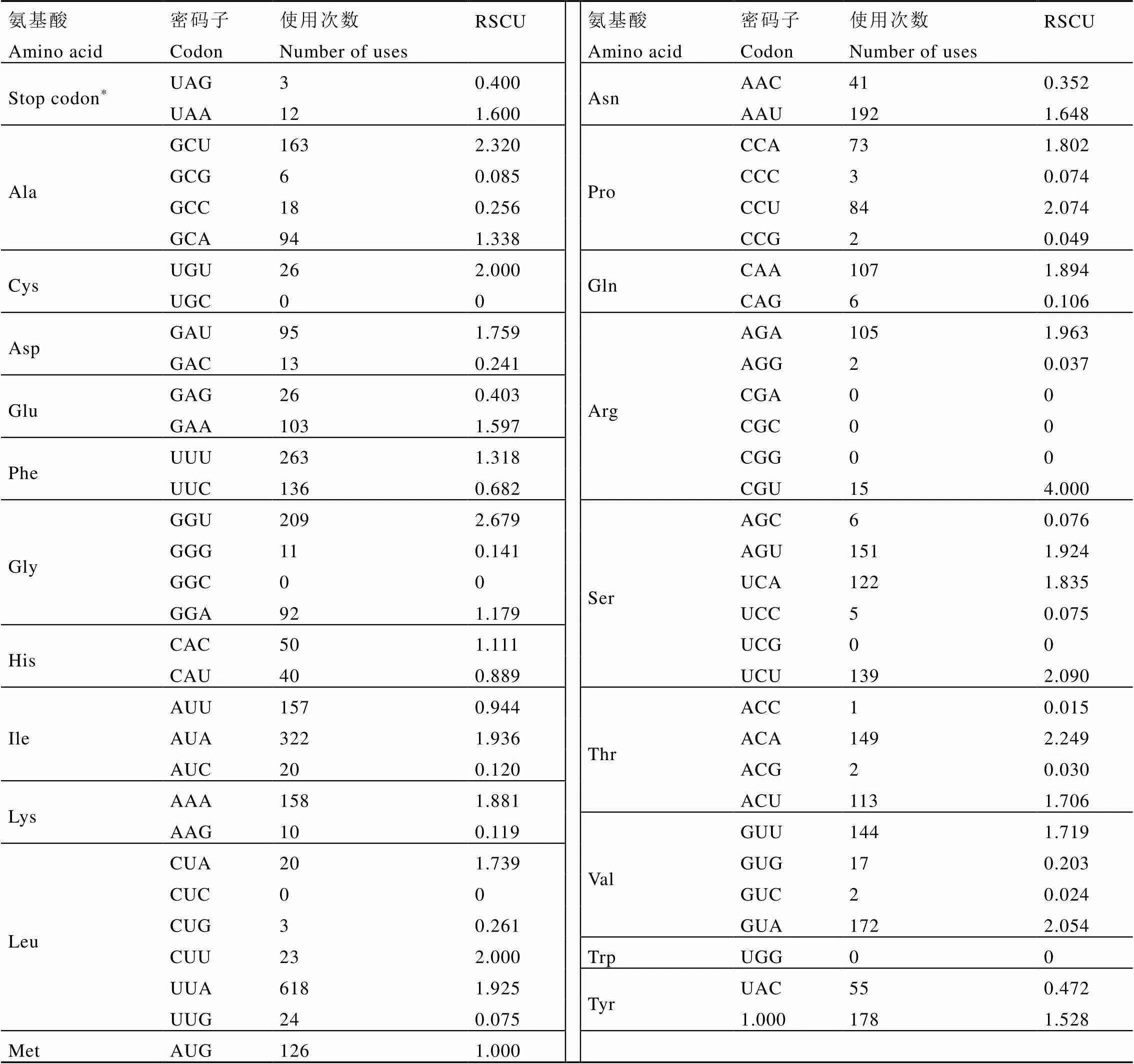
表4 桔青霉线粒体基因组蛋白质编码基因密码子使用情况
2.5 线粒体基因组tRNA基因
桔青霉线粒体基因组包括24个tRNA基因,其二级结构如图4所示,tRNA基因长度较为均一,介于71~86 bp。24个tRNA均包含有氨基酸受体臂、双氢尿嘧啶臂(DHU臂)、反密码子臂及TΨC臂,可形成典型的三叶草结构。24个tRNA中,除tRNAHis-GTG与tRNAArg-ACG氨基酸受体臂长度为6 bp外,其余氨基酸受体臂长度均为7 bp。反密码子臂中只有tRNAGlu-TTC、tRNAVal-TAC、tRNAMet-CAT和tRNALys-TTT的臂长为4 bp,其余均为5 bp。所有tRNA的反密码子环和TΨC环长度均为7 bp。此外,共有5个tRNA(tRNALeu-TAA、tRNALeu-TAG、tRNATyr-GTA、tRNASer-GCT、tRNASer-TGA)存在可变环。
tRNA在二级结构折叠过程中,会出现碱基错配情况,全部tRNA中共出现30处碱基错配,均属于G-U错配。错配碱基对在tRNA分子的不同部位均有分布:氨基酸受体臂13对、反密码子臂7对、TΨC臂5对、双氢尿嘧啶臂4对、可变臂1对,此类G-U碱基错配现象在近缘种微生物的tRNA二级结构中也较为常见[28]。
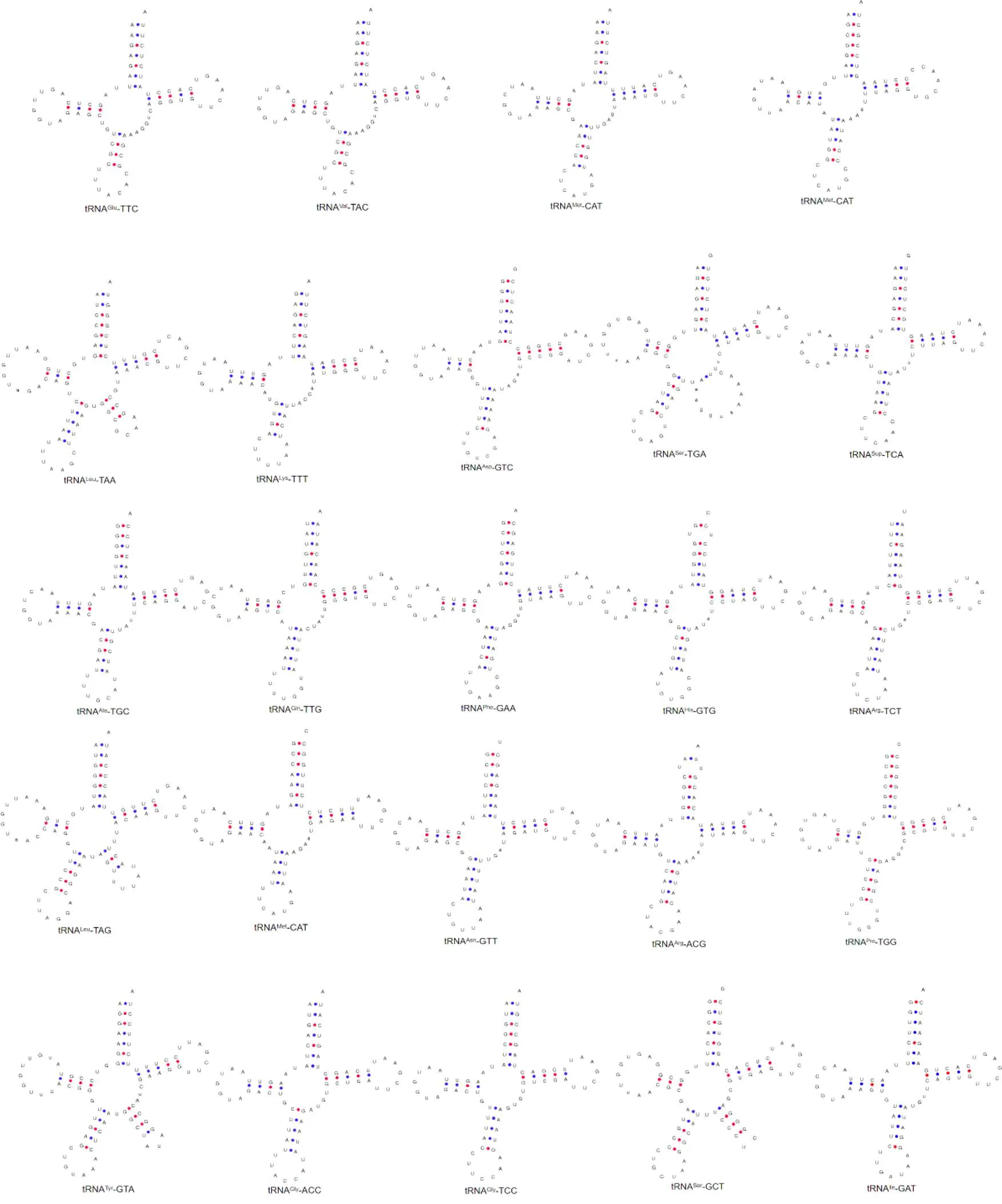
图4 桔青霉线粒体基因组tRNA二级结构预测图
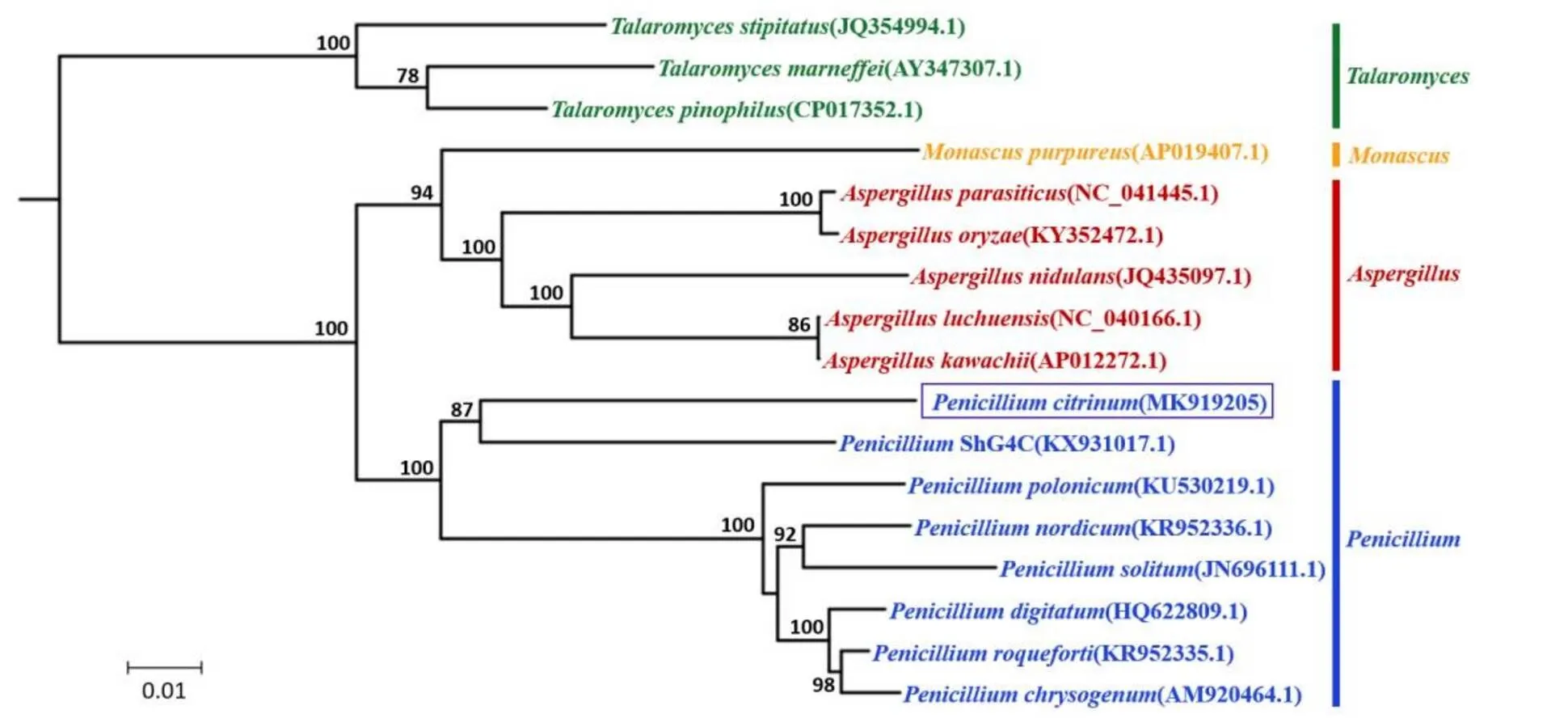
图5 基于17种真菌线粒体蛋白质编码基因序列构建的Maximum likelihood系统进化树
2.6 线粒体基因组微卫星序列
根据IMEX得到的结果,桔青霉线粒体微卫星序列共有249个,所有的微卫星序列总长度为1 930 bp,约占基因组全长的7%,计算得到的相对丰度和相对密度分别为9.04和70.09,表示每1 000个碱基中平均有9.04个微卫星序列,每1 000个碱基中微卫星序列的平均长度为70.09 bp。统计结果显示,一型微卫星91个、二型微卫星121个、三型微卫星31个、四型微卫星5个、五型微卫星1个,其中二型微卫星占比最高,且微卫星长度总体较短,推测是由于线粒体基因组微卫星序列突变率较高,造成基序单位的丢失或突变,从而形成短的微卫星序列[29]。
2.7 桔青霉近缘种及系统发育地位
为研究桔青霉和近缘物种的关系,以、、作为外群,基于17种微生物的线粒体蛋白质编码基因核苷酸序列构建了系统发育树(图5)。由结果可知,属与属的亲缘关系最为接近,其次是属。这一结果与Joardar等[24]以及Kang等[30]的研究一致。在青霉属中,与桔青霉亲缘关系最近的是ShG4C,二者单独聚为一支,其次是与。此外,本研究基于17种真菌线粒体蛋白编码基因序列,采用Maximum likelihood法构建的系统发育树与类似研究的结果较为接近,如与亲缘关系非常接近[27];则是与聚为一支[26],说明了线粒体基因组信息能较为稳定、可靠的反映不同微生物的种间关系。
此外,由于线粒体基因组进化速度快于物种进化速度,环境因素以及生物行为习性会更大程度的影响其DNA进化,可能导致基因出现定向的改变,使区域种群间出现不同程度的遗传差异[31-32]。因此,线粒体基因组信息不仅能一定程度反映物种的遗传背景[33-34],甚至可以作为划分其地理种群的依据[35],在分子水平上亲缘关系较近的生物通常分布在邻近的区域或相似的生境中。除桔青霉外,其近缘种也常在茶叶相关的生境中被检测到,如从云南普洱熟茶中检测到草酸青霉()、灰黄青霉()、青霉变种()、产黄青霉()、棘孢青霉()[36];在茯砖茶发花过程中检测到斜卧青霉()[37];在茶叶种植园的土壤中分离出鲜红青霉()、产黄青霉()、顶青霉()、蓝青霉()、斜卧青霉()、红色青霉()等[38],进一步说明青霉属微生物在茶制品及其栽培环境中广泛存在。
3 讨论
本研究首次对黑茶中桔青霉线粒体全基因组进行测定与分析,其序列全长为27 537 bp,包括15个蛋白质编码基因、2个rRNA、24个tRNA以及1个独立的ORF。不同类型基因A(36.17%)、T(37.06%)、C(11.82%)、G(14.95%)碱基的含量比例与已报道的近缘种微生物比例接近,基因组排列顺序符合青霉菌的典型特征。桔青霉线粒体基因组AT偏斜为–0.012 2,表现出弱AT负偏移,同样表现出弱AT负偏移的还有系统发育关系上与之最接近的ShG4C(–0.002 2),其他青霉属物种则没有出现这种现象,根据Weber等[39]的研究结果可知,AT偏斜与物种生活的地理位置及环境温度存在一定关联,推测桔青霉与ShG4C在生长环境选择上有一定的相似性。
桔青霉15个蛋白质编码基因可分为编码ATP合成酶亚基、编码细胞色素、编码氧化还原酶亚基与核糖体蛋白编码基因,均使用典型的起始密码子ATG与终止密码子TAA/TAG。蛋白质编码基因编码最多的氨基酸依次为Leu(14.55%)、Ile(10.56%)、Ser(8.95%)和Phe(8.44%);RSCU频率较高的4个密码子分别是UUA(618次)、AUA(322次)、UUU(263次)和GGU(209次),与近缘种同样表现出对A和U碱基的偏好性。24个tRNA长度在71~86 bp,均可形成稳定的三叶草型结构,tRNA碱基对上存在30处G-U错配,错配碱基在二级结构的不同部位上均有分布。线粒体基因组共包含有249个微卫星序列,其中以长度较短的一、二型微卫星序列为主,推测是由于基因组微卫星序列突变率较高,造成基序单位的丢失或突变所致。桔青霉线粒体基因组信息显示,其具有典型的青霉属物种特征,在进化上较为保守。
系统发育树分析结果表明,与属微生物亲缘关系最接近的是属,这与真菌分类学的观点一致。在青霉属内,桔青霉与ShG4C的亲缘关系最为接近,二者在系统发育关系上单独聚为一小支,进一步参考Mardanov等[26]的研究可知,ShG4C与桔青霉在线粒体基因组核苷酸组成、基因排列上同样非常相近,鉴于此,本文推测桔青霉与ShG4C遗传背景相似且出现分化的时间较晚。此外,与桔青霉系统发育地位较接近的微生物还有与,参照Khalil等[40]、Iacumin等[41]的研究,这2株真菌分别由霉变的橄榄与培根内分离获得,且与桔青霉在形态学上有一定的相似性。
本研究从菌种形态和线粒体信息方面对桔青霉进行了描述,为明确该菌株的分类地位提供了重要的依据,也为后续进一步对青霉属物种进行线粒体基因组测定和分析,明确近缘种之间的系统发育关系提供了基础。此外,通过分子生物学技术对黑茶发酵过程中微生物种群实施监测,能更好的指导茶叶精控发酵工作的开展。如何控制桔青霉这一菌株发达的蛋白酶系在茶叶渥堆中发挥作用,而在后续工序中不再繁殖,是提升黑茶品质及保障微生物安全的可行思路。目前,由于青霉属已进行了线粒体基因组测序的物种仍有限,要更加准确的界定青霉属下354个接受种[42]分类地位及其相互之间的系统发育关系,还有待更多的测序数据支持。
[1] Desmond E, Brochier-Armanet C, Forterre P, et al. On the last common ancestor and early evolution of eukaryotes: reconstructing the history of mitochondrial ribosomes [J]. Research in Microbiology, 2011, 162(1): 53-70.
[2] Hua Y Q, Yan Z T, Fu W B, et al. Sequencing and analysis of the complete mitochondrial genome in(Diptera: Culicidae) [J]. Mitochondrial DNA Part A, 2015, 27(4): 2909-2910.
[3] Houbraken J A M P, Frisvad J C, Samson R A. Taxonomy ofand related species [J]. Fungal Diversity, 2010, 44(1): 117-133.
[4] Haas D, Pfeifer B, Reiterich C, et al. Identification and quantification of fungi and mycotoxins from Pu-erh tea [J]. International Journal of Food Microbiology, 2013, 166(2): 316-322.
[5] 粟清. 真菌单菌株固态发酵茶叶及其产Teadenol化合物分析[D]. 昆明: 云南大学, 2018. Su Q. Fungal solid-state fermentation teas and its teadenol-producing potentiality [D]. Kunming: Yunnan University, 2018.
[6] 赵仁亮, 谭吉慧, 卢秦华, 等. 茯砖茶发花微生物生物学特性研究[J]. 茶叶科学, 2016, 36(2): 160-168. Zhao R L, Tan J H, Lu Q H, et al. Biological characterization of fungi involved in fu brick tea fermentation [J]. Journal of Tea Science, 2016, 36(2): 160-168.
[7] 熊元元. 四川黑茶渥堆微生物多样性及空气微生物研究[D]. 雅安: 四川农业大学, 2017. Xiong Y Y. Study on Microbial diversity of Sichuan dark tea during post-fermentation and airborne microbial [D]. Ya'an: Sichuan Agricultural University, 2017.
[8] 姜依何, 胥伟, 朱旗. 黑茶真菌污染研究进展及探讨[J]. 茶叶科学, 2018, 38(3): 227-236. Jiang Y H, Xu W, Zhu Q. Research progress and discussion on fungal contamination of dark tea [J]. Journal of Tea Science, 2018, 38(3): 227-236.
[9] Li T T, Jiang G X, Qu H X, et al. Comparative transcriptome analysis ofcultured with different carbon sources identifies genes involved in citrinin biosynthesis [J]. Toxins, 2017, 9(2): 69. doi: 10.3390/toxins9020069.
[10] Föllmann W, Behm C, Degen G H. Toxicity of the mycotoxin citrinin and its metabolite dihydrocitrinone and of mixtures of citrinin and ochratoxin A[J]. Archives of Toxicology, 2014, 88(5): 1097-1107.
[11] 赵兴丽, 张金峰, 周玉锋, 等. 一株拮抗茶炭疽病菌的木霉菌的分离、筛选及鉴定[J]. 茶叶科学, 2019, 39(4): 431-439. Zhao X L, Zhang J F, Zhou Y F, et al. Isolation, Screening and identification of a strain of trichoderma antagonizing tea anthracnose [J]. Journal of Tea Science, 2019, 39(4): 431-439.
[12] Wu L, Zhao Y L, Xu Z G, et al. The complete mitochondrial genome and phylogeny of(Passeriformes: Thraupidae) [J]. Conservation Genetics Resources, 2019, 11: 191-193.
[13] Patel R K, Mukesh J. NGS QC Toolkit: a toolkit for quality control of next generation sequencing data [J]. Plos One, 2012, 7(2): e30619. doi: 10.1371/journal.pone.0030619.
[14] Peng Y, Leung H C M, Yiu S M, et al. IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth [J]. Bioinformatics, 2012, 28(11): 1420-1428.
[15] Donath A, Jühling F, Externbrink F, et al. MITOS: Improved de novo metazoan mitochondrial genome annotation [J]. Molecular Phylogenetics and Evolution, 2013, 69(2): 313-319.
[16] Lohse M, Drechsel O, Bock R. OrganellarGenomeDRAW (OGDRAW): a tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes [J]. Current Genetics, 2007, 52: 267-274.
[17] Lowe T M, Eddy S R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence [J]. Nucleic Acids Research, 1997, 25(5): 955-964.
[18] Rasmus W. FeatureExtract-extraction of sequence annotation made easy [J]. Nucleic Acids Research, 2005, 33(s2): 567-569.
[19] Xia X H, Xie Z. DAMBE: software package for data analysis in molecular biology and evolution [J]. Journal of Heredity, 2001, 92(4): 371-373.
[20] 周思倩, 焦伟丽, 彭珠黎, 等. 埃博拉病毒基因组中微卫星序列的分布分析[J]. 基因组学与应用生物学, 2019, 38(3): 1087-1095. Zhou S Q, Jiao W L, Peng Z L, et al. Analysis of microsatellite sequence distribution in Ebolavirus genomes [J]. Genomics and Applied Biology, 2019, 38(3): 1087-1095.
[21] 彭艳, 陈斌, 李廷景. 黄侧异腹胡蜂线粒体基因组全序列测定和分析[J]. 昆虫学报, 2017, 60(4): 464-474. Peng Y, Chen B, Li T J. Sequencing and analysis of the complete mitochondrial genome of(Hymenoptera: Vespidae) [J]. Acta Entomologica Sinica, 2017, 60(4): 464-474.
[22] 孔华忠. 中国真菌志(第三十五卷): 青霉属及其相关有性型属[M]. 北京: 科学出版社, 2007: 121-124. Kong H Z. Mycolography of China(vol.35): Penicillium and its related sex type [M]. Beijing: Science Press, 2007: 121-124.
[23] Hu Z Y, Liu S Q, Xu Z G, et al. Complete mitochondrial genome and phylogenetic analysis ofin dark tea [J]. Mitochondrial DNA Part B, 2019, 4(2): 2445-2446.
[24] Joardar V, Abrams N F, Hostetler J, et al. Sequencing of mitochondrial genomes of nineandspecies identifies mobile introns and accessory genes as main sources of genome size variability [J]. BMC Genomics, 2012, 13(1): 698. doi: 10.1186/1471-2164-13-698
[25] Eldarov M A, Mardanov A V, Beletsky A V, et al. Complete mitochondrial genome of compactin-producing fungussolitum and comparative analysis ofmitochondrial genomes [J]. FEMS Microbiology Letters, 2012, 329(1): 9-17.
[26] Mardanov A V, Glukhova L B, Gruzdev E V, et al. The complete mitochondrial genome of the acid-tolerant fungusShG4C [J]. Genomics Data, 2016, 10: 141-143.
[27] Sun X, Li H, Yu D. Complete mitochondrial genome sequence of the phytopathogenic fungusand comparative analysis of closely related species [J]. Fems Microbiology Letters, 2011, 323(1): 29-34.
[28] Woo P, Zhen H, Cai J, et al. The mitochondrial genome of the thermal dimorphic fungusis more closely related to those of molds than yeasts [J]. Febs Letters, 2003, 555(3): 469-477.
[29] Ellegren H. Microsatellites: simple sequences with complex evolution [J]. Nature Reviews Genetics, 2004, 5(6): 435-445.
[30] Kang X, Liu C, Liu D, et al. The complete mitochondrial genome of huperzine A-producing endophytic fungus[J]. Mitochondrial Dna Part B Resources, 2016, 1(1): 202-203.
[31] Avise J C. The history and purview of phylogeography: a personal reflection [J]. Molecular Ecology, 1998, 7(4): 371-379.
[32] 李宏俊, 张晶晶, 袁秀堂, 等. 利用线粒体COI和微卫星标记分析文蛤7个地理群体的遗传变异[J]. 生态学报, 2016, 36(2): 499-507. Li H J, Zhang J J, Yuan X T, et al. Genetic diversity and differentiation of seven geographical populations of hard clam () assessed by COI and microsatellite markers [J]. Acta Ecologica Sinica, 2016, 36(2): 499-507.
[33] 连总强, 滚双宝, 李力, 等. 基于第二代测序技术兰州鲇线粒体基因组全序列测定与分析[J]. 水生生物学报, 2017, 41(2): 334-345. Lian Z Q, Gun S B, Li L, et al. Sequencing and analysis of the complete mitochondrial genome ofbased on next generation sequencing technologies [J]. Acta Hydrobiologica Sinica, 2017, 41(2): 334-345.
[34] 刘小丽, 孙佼, 韩金巧, 等. 岛屿生境下黄毛鼠种群的遗传变异[J]. 生态学报, 2019, 39(18): 6898-6907. Liu X L, Sun J, Han J Q, et al. Genetic variation of Rattus losea populations in island habitats [J]. Acta Ecologica Sinica, 2019, 39(18): 6898-6907.
[35] Morin P A, Archer F I, Foote A D, et al. Complete mitochondrial genome phylogeographic analysis of killer whales () indicates multiple species [J]. Genome Research, 2010, 20(7): 908-916.
[36] 赵振军, 童华荣, 周黎, 等. 普洱茶中真菌种群的分离与分子鉴定[J]. 茶叶科学, 2009, 29(6): 436-442. Zhao Z J, Tong H R, Zhou L, et al. Isolation and molecular identification of fungal colonization of Pu-erh tea [J]. Journal of Tea Science, 2009, 29(6): 436-442.
[37] 刘石泉, 赵运林, 胡治远. DGGE法初步解析茯砖茶发花过程中真菌群落结构[J]. 生态学杂志, 2014, 33(10): 2687-2693. Liu S Q, Zhao Y L, Hu Z Y. Analysis of fungal community structure during the Fahua-fermentation process of Fuzhuan Brick Tea by DGGE technology [J]. Chinese Journal of Ecology, 2014, 33(10): 2687-2693.
[38] Karaoglu S A, Ulker S. Isolation, identification and seasonal distribution of soilborne fungi in tea growing areas of Iyidere-Ikizdere vicinity (Rize-Turkey) [J]. Journal of Basic Microbiology, 2006, 46(3): 208-218.
[39] Weber C C, Hurst L D. Intronic AT skew is a defendable proxy for germline transcription but does not predict crossing-over or protein evolution rates in[J]. Journal of Molecular Evolution, 2010, 71(5-6): 415-426.
[40] Khalil A M A, Hashem A H, Abdelaziz A M. Occurrence of toxigenicin retail green table olives from the Saudi Arabia market [J]. Biocatalysis and Agricultural Biotechnology, 2019, 21: 101314. doi: 10.1016/j.bcab.2019.101314
[41] Iacumin L, Manzano M, Andyanto D, et al. Biocontrol of ochratoxigenic moulds (and) byandduring speck production [J]. Food Microbiology, 2017, 62: 188-195.
[42] Visagie C M, Houbraken J, Frisvad J C, et al. Identification and nomenclature of the genus[J]. Studies in Mycology, 2014, 78: 343-371.
Sequencing and Analysis of the Complete Mitochondrial Genome ofin Hunan Yiyang Dark Tea
HU Zhiyuan1,2, LIU Suchun1*, XU Zhenggang2, LIU Shiquan2, WEN Xin3
1. College of Food Science and Technology, Hunan Agricultural University, Changsha 410128, China; 2. School of Materials and Chemical Engineering, Hunan City University, Yiyang 413000, China; 3. Hunan Institute of Food Quality Supervision Inspection and Research, Changsha 410111, China
In this study, the mitochondrial genome sequence of a strain ofisolated from dark tea was determined and analyzed, and its phylogenetic relationship with the closely related microorganisms was explored. The result shows that the mitochondrial genome ofis a circular DNA molecule with a length of 27 537 bp, which encodes 42 genes. The genome bases are composed of A (36.17%), T (37.06%), C (11.82%) and G (14.95%). All the 15 protein-coding genes use typical ATG as the start codon, TAA or TAG as the stop codon. Its gene sequences are similar to those of the reportedspecies and are conserved in evolution. The highly occurred amino acids in the protein-coding genes are Leu, Ile, Ser and Phe. The top 4 codons with the high RSCU frequency are UUA, AUA, UUU and GGU, respectively. There are 30 G-U mismatches in 24 tRNA genes, all of which could form typical cloverleaf structure. Phylogenetic analysis shows that the most closely related taxonomic status ofisShG4C, followed byand.
dark tea,, mitochondrial genome, phylogeny
S571.1;Q939.5
A
1000-369X(2020)06-830-15
2020-01-16
2020-03-30
湖南省黑茶金花重点实验室资助项目(湘科规财[2019]8号)、湖南省重点研发计划资助项目(2018NK2036)、安化黑茶非遗传承湖南省社会科学普及基地(湘社普[2019]6号)
胡治远,男,讲师,博士研究生,主要从事茶及天然植物的开发与利用研究。*通信作者:liusc@hunau.net
猜你喜欢
特产研究(2022年6期)2023-01-17 05:05:06
教学考试(高考生物)(2020年6期)2020-11-23 05:25:56
生物学通报(2020年11期)2020-10-22 01:20:20
食品与生物技术学报(2020年8期)2020-01-06 08:00:56
科学24小时(2019年5期)2019-06-11 08:39:38
发明与创新(2019年9期)2019-03-26 02:22:48
中成药(2018年7期)2018-08-04 06:04:10
四川动物(2017年4期)2017-07-31 23:54:19
集美大学学报(自然科学版)(2015年4期)2015-02-28 01:13:37
河北遥感(2014年3期)2014-07-10 13:16:48
