
二甲双胍通过激活AMPK抑制肺动脉高压大鼠的肺血管重塑
2020-12-10 08:48:38尚文丽张莹莹陈芬芬王贵佐陕西省人民医院呼吸与危重症医学科西安70068西安交通大学第二附属医院呼吸与危重症医学科西安交通大学第二附属医院干部保健办公室通信作者mailsesoryyeahnet
山西医科大学学报 2020年11期
尚文丽,张莹莹,陈芬芬,王贵佐,韩 冬(陕西省人民医院呼吸与危重症医学科,西安 70068;西安交通大学第二附属医院呼吸与危重症医学科;西安交通大学第二附属医院干部保健办公室;通信作者,E-mail:sesory@yeah.net)
肺动脉高压(pulmonary artery hypertension,PAH)是由多种因素引起的肺血管功能及结构性异常,导致肺血管阻力增加,引起肺动脉压力升高的临床综合征,严重者可导致右心衰竭甚至死亡[1]。PAH的发病机制包括:肺血管紧张性增高、肺血管重塑和原位血栓形成[2]。肺血管重塑是PAH的主要病理学基础,目前普遍认为肺动脉平滑肌细胞(pulmonary arterial smooth muscle cells,PASMCs)的增殖及其向内膜下的迁移是肺血管重塑的主要病理机制[3]。因此,阐明PASMCs异常增殖的分子生物学机制及寻找相关的干预措施,明确有效的治疗靶点是目前PAH治疗的关键。
腺苷酸活化蛋白激酶(AMP-activated protein kinase,AMPK)是一种广泛参与人体内能量代谢活动的重要蛋白激酶[4],其常见激动剂有二甲双胍及AICAR等。研究发现,激活AMPK可抑制野百合碱诱导的大鼠PAH模型的发生[5],其作用机制尚待研究。一项人头颈部鳞癌细胞中的研究[6]发现,二甲双胍可剂量依赖性地抑制癌细胞系的生长。与此同时,二甲双胍上调了磷酸化AMPK的蛋白水平并下调了S期蛋白相关激酶2(S phase kinase associated protein 2,Skp2)的表达。另一项小鼠主动脉血管平滑肌细胞中的研究发现,敲除AMPKα2可上调Skp2的表达并下调P27的蛋白水平[7]。然而激活AMPK对大鼠PAH模型发生的抑制作用是否通过调控Skp2/P27信号通路尚不明确。
在本研究中,我们应用野百合碱诱导的PAH大鼠模型,评估二甲双胍激活AMPK信号通路抑制PAH模型发生的可能机制,为肺动脉高压治疗提供新的思路与方法。
1 材料与方法
1.1 实验动物与主要试剂
选用西安交通大学医学院实验动物中心提供的4周龄近交系雄性SD大鼠30只,体质量在200-250 g之间。二甲双胍(格华止,国药准字H20023371),由上海施贵宝制药有限公司提供。野百合碱(monocrotaline,MCT)购于美国Sigma-Aldrich公司。兔抗鼠AMPK、p-AMPK、Skp2、P27、GAPDH一抗购于美国Cell Signaling Technology公司,HRP偶联的羊抗兔抗体由美国Sigma-Aldrich公司提供,RIPA裂解液由武汉博士德生物工程有限公司提供。
1.2 实验分组和模型构建
将4周龄体质量200-250 g的30只雄性SD大鼠随机分为三组:对照组、PAH组和PAH+二甲双胍治疗组(简称为二甲双胍组)。大鼠适应性饲养3 d后,PAH组按60 mg/kg腹腔注射野百合碱(Day 1);二甲双胍组按60 mg/kg腹腔注射野百合碱(Day 1),并按150 mg/(kg·d)腹腔注射二甲双胍(Day 1-28);对照组腹腔注射等量生理盐水(Day 1-28)。大鼠饲养于西安交通大学医学院实验动物中心清洁级动物房。
1.3 右心室收缩压测定
通过血流动力学指标检测确定PAH大鼠模型构建是否成功,10%水合氯醛(0.3 ml/kg)腹腔注射麻醉大鼠,从右侧颈外静脉插入微导管,通过压力传感器及多导生理记录仪测定大鼠右心室收缩压(right ventricle systolic pressure,RVSP),待压力波形图平稳后记录三组数据,计算平均值。
1.4 右心室肥大指数测定
将心脏去除左、右心房,沿室间隔(interventricular septum,S)分离左、右心室,生理盐水冲洗后滤纸吸干,分别称量右心室(right ventricular,RV)、室间隔加左心室(left ventricular,LV)质量,计算右心肥大指数[RV/(LV+S)]。
1.5 肺脏苏木素-伊红染色(HE染色)观察肺血管病理学
取出分离后的左肺组织利用10%中性缓冲甲醛液固定,石蜡包埋,切片(6 μm),苏木精-伊红(HE)染色,观察肺血管病理学变化。
1.6 免疫印迹法检测AMPK、p-AMPK、Skp2及P27
精密称取各组大鼠肺组织,根据RIPA裂解液使用说明书制备肺组织匀浆,肺组织与RIPA裂解液的比例为1 mg ∶7.5 μl,用玻璃匀浆器上下、旋转充分碾磨。在冰上裂解1 h后,14 000 r/min离心20 min,取上清,利用Western blot检测各组大鼠肺组织Skp2(稀释度1 ∶1 000)及P27(稀释度1 ∶1 000)表达、AMPK(稀释度1 ∶500)及p-AMPK(稀释度1 ∶500)变化,Skp2及P27表达应用GAPDH(稀释度1 ∶5 000)作为内参校正。Image Lab图像分析软件进行条带灰度值的测量分析。
1.7 统计学分析
2 结果
2.1 各组大鼠一般状况观察
对照组大鼠食量稳定,皮毛光泽,呼吸、心跳平稳,活动自如,体质量明显增加,无死亡;PAH组大鼠食量减少,皮毛粗糙,活动减少,体质量增加缓慢,4周内总计死亡4只;二甲双胍组大鼠与PAH组相比,食量有所增加,毛发稍光滑,活动增多,体质量较PAH组增加,状态明显好于PAH组,且大鼠无死亡。
2.2 右心室收缩压及右心室肥大指数
造模4周后测定大鼠右心室收缩压(RVSP)及计算各组大鼠右心肥大指数[RV/(LV+S)]。PAH组RVSP较对照组明显增高,差异具有统计学意义[(45.88±3.45)mmHgvs(22.36±2.19)mmHg,P<0.05,见图1],提示肺动脉高压模型建立成功;而二甲双胍组RVSP[(29.81±1.97)mmHg]较PAH组显著下降(P<0.05,见图1),提示二甲双胍可显著降低野百合碱诱导的肺动脉压力增高。同样与对照组相比,PAH组RV/(LV+S)显著增加[(58.97±3.62)%vs(15.94±0.87)%,P<0.05,见图1],提示PAH组大鼠右心室明显肥厚;而二甲双胍组RV/(LV+S)[(32.80±3.05)%]较PAH组则显著降低(P<0.05,见图1),提示二甲双胍可显著抑制右心室重构的发生。
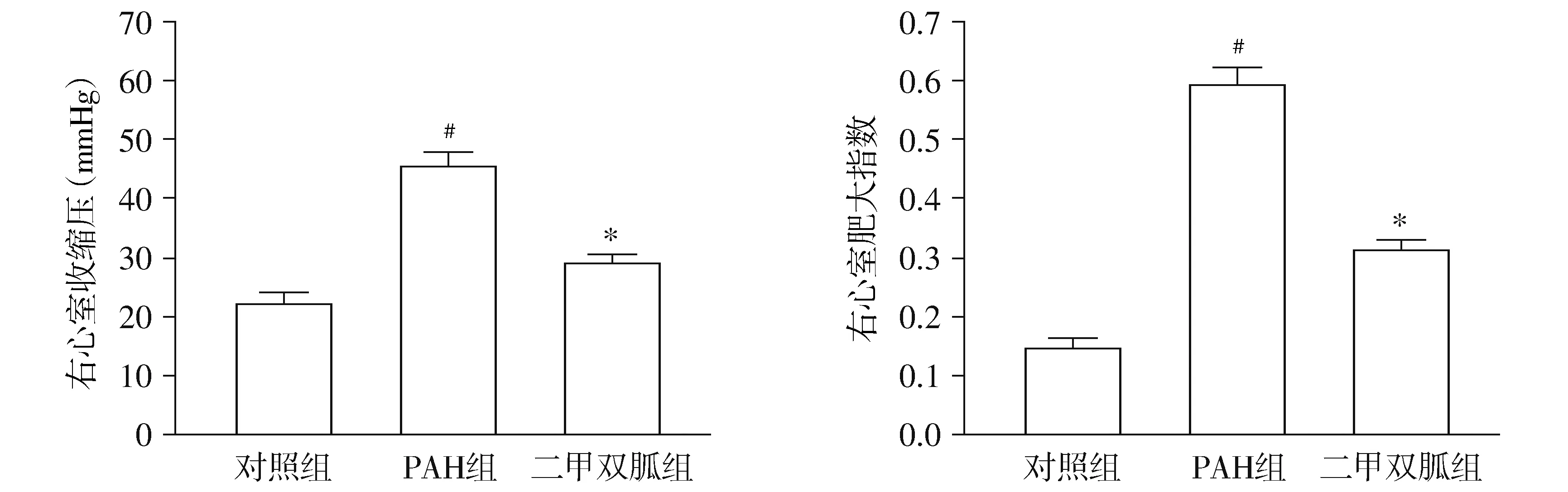
与对照组比较,#P<0.05;与PAH组比较,*P<0.05图1 二甲双胍抑制野百合碱诱导的大鼠肺动脉高压模型的发生Figure 1 Metformin prevents against MCT-induced PAH
2.3 肺血管病理学HE染色
各组大鼠的肺血管病理形态学改变如下:光镜下可见对照组大鼠肺小动脉管壁厚薄较一致,形态规则,血管腔内壁及外周均未见炎细胞浸润(见图2)。而PAH组大鼠肺小动脉管壁明显增厚,管腔狭窄严重,内膜处可见大量炎性细胞浸润;肺血管损伤明显,大量的血管内皮细胞肿胀、变性,部分已突向血管腔内,甚至出现坏死、脱落(见图2)。二甲双胍组肺小动脉管壁增厚较PAH组明显改善,其炎细胞浸润及内皮损伤亦较PAH组减轻(见图2)。
2.4 总AMPK及磷酸化AMPK蛋白的水平
Western blot结果显示,三组的T-AMPK蛋白水平无明显差异(见图3);PAH组p-AMPK蛋白水平较对照组显著降低,差异具有统计学意义(P<0.05);而二甲双胍组的p-AMPK蛋白水平与对照组相比无显著差异,而较PAH组显著升高(P<0.05,见图3)。
2.5 二甲双胍抑制野百合碱诱发的Skp2表达增高
Western blot结果显示,PAH组Skp2蛋白表达水平较对照组显著增高,差异具有统计学意义(P<0.05);而二甲双胍组Skp2蛋白水平较PAH组显著降低,差异具有统计学意义(P<0.05,见图4)。
2.6 二甲双胍逆转野百合碱诱导的P27表达减低
Western blot结果显示,PAH组P27表达水平较对照组显著降低,差异具有统计学意义(P<0.05);而二甲双胍组P27蛋白水平较PAH组显著增高,差异具有统计学意义(P<0.05,见图5)。
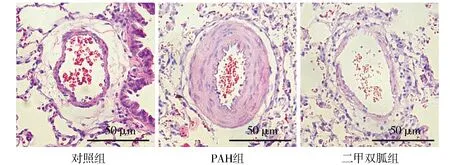
图2 各组大鼠肺小动脉HE染色 (×200)Figure 2 HE-staining of small pulmonary vessels in rats (×200)
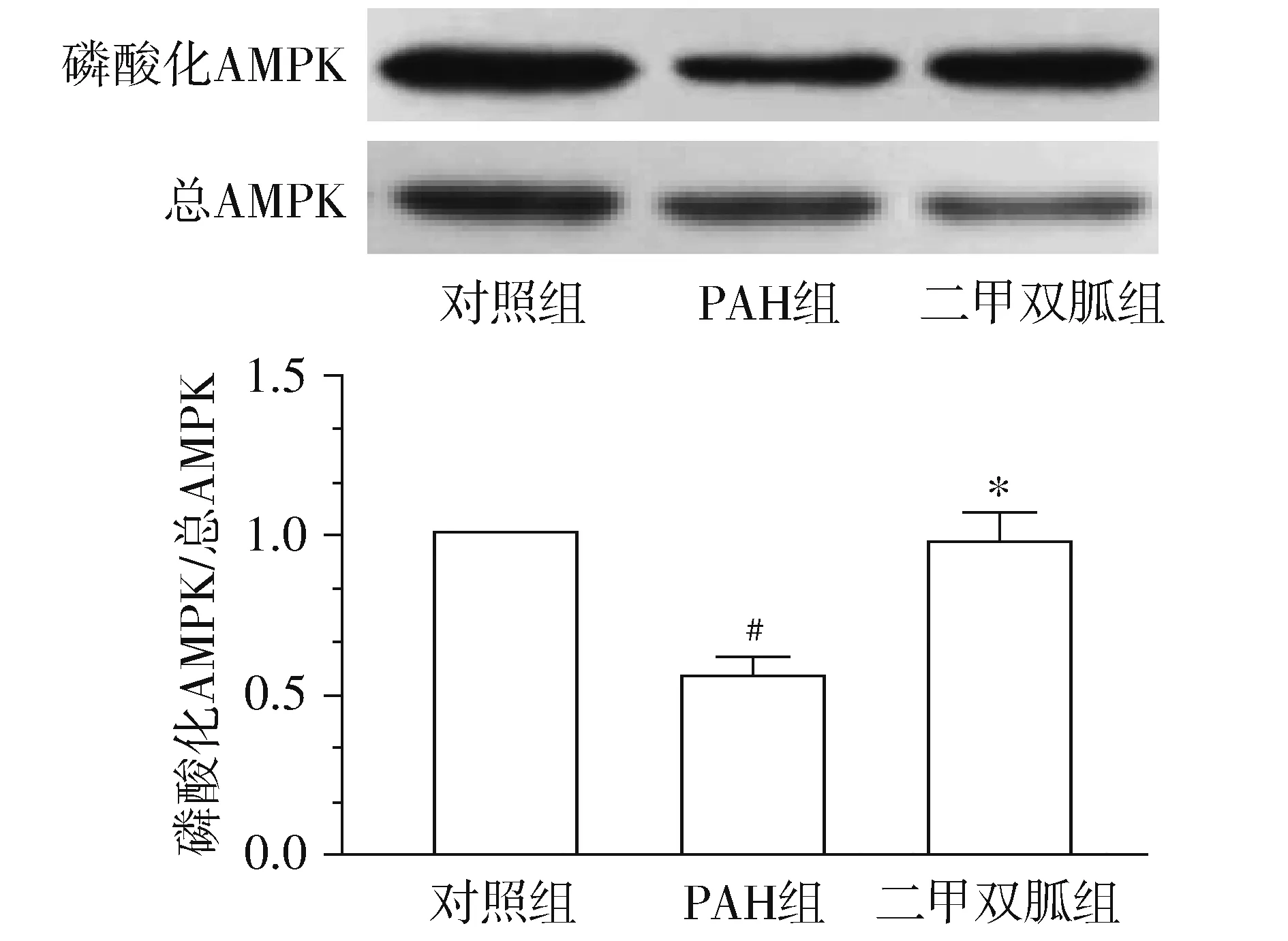
与对照组比较,#P<0.05;与PAH组比较,*P<0.05图3 Western blot检测二甲双胍对肺组织内AMPK磷酸化水平的影响Figure 3 Effects of metformin on the phosphorylation of AMPK in rat lung tissues by Western blot
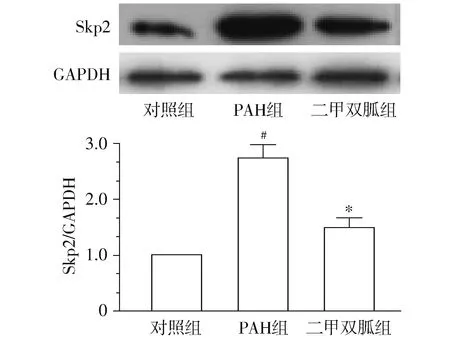
与对照组比较,#P<0.05;与PAH组比较,*P<0.05图4 Western blot检测二甲双胍对肺组织内Skp2表达的影响Figure 4 Effects of metformin on the expression of Skp2 protein in rat pulmonary tissues by Western blot
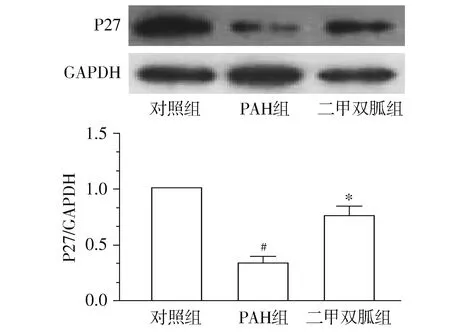
与对照组比较,#P<0.05;与PAH组比较,*P<0.05图5 Western blot检测二甲双胍对肺组织内P27表达的影响Figure 5 Effects of metformin on the expression of P27 protein in rat pulmonary tissues by Western blot
3 讨论
细胞增殖与细胞增殖周期相关,而细胞增殖周期受细胞周期蛋白(cyclin)及其依赖蛋白激酶(cyclin-dependent kinase,CDK)和CDK抑制物(CDK inhibitor,CDKI)的共同调控。P27是抑制细胞增殖的重要因子之一[8]。近年来动物模型和细胞层面的研究发现,PAH时P27是肺动脉平滑肌细胞(pulmonary arterial smooth muscle cells,PASMCs)中唯一表达量显著减少的CDKI,上调P27可抑制PASMC增殖,抑制肺血管重塑和肺动脉高压的发生[9-11]。Skp2是P27泛素化降解过程中的一个重要调控分子,其过表达可下调P27的蛋白水平[12,13]。
Gao等[9]研究发现,阿托伐他汀可降低左肺全切并野百合碱诱导的肺动脉高压大鼠模型的mPAP和右室肥大指数,并可上调P27的表达。Liu等[10]研究发现法舒地尔可通过上调P27的表达,抑制血小板衍生生长因子(platelet-derived growth factor,PDGF)诱导的人肺动脉平滑肌细胞(human pulmonary arterial smooth muscle cells,HPASMCs)增殖。另一项研究[11]发现,PPARδ的特异性配体GW501516,可通过激活PPARδ上调P27的表达,并显著抑制PDGF诱导的人肺动脉平滑肌细胞增殖。以上这些研究表明,通过不同途径上调P27的表达,可抑制体外培养的PASMCs的过度增殖,并抑制PAH动物模型中肺血管重塑和肺动脉高压的发生。
被药理学方法激活的AMPK,可抑制Skp2介导的P27的降解,上调P27的表达。Song等[7]在一项小鼠主动脉平滑肌细胞中的研究发现,敲除AMPKα2可上调Skp2的表达并下调P27的水平。另一项人头颈部鳞癌细胞中的研究发现,二甲双胍可上调磷酸化AMPK的蛋白水平并下调Skp2的表达,剂量依赖性地抑制癌细胞系的生长[6]。本研究中,野百合碱诱发的PAH大鼠模型中肺组织Skp2蛋白水平显著升高,P27蛋白水平明显降低,提示Skp2/P27信号通路的失衡参与了肺动脉高压的发生。模型动物经AMPK激动剂二甲双胍治疗后,肺组织中AMPK磷酸化水平显著升高,Skp2的过表达被抑制,P27蛋白水平基本恢复正常。提示激活AMPK可抑制Skp2的过表达,减少P27的泛素化降解,从而抑制肺动脉平滑肌细胞的过度增殖,进一步抑制肺血管重塑的发生。
本研究中我们发现,在野百合碱诱发的PAH大鼠模型中,存在着明显的肺血管重塑,表现为肺血管壁明显增厚,中膜平滑肌细胞层增厚,导致肺小动脉管腔严重狭窄甚至阻塞。血流动力学检测提示肺动脉压力显著增高;右心室质量增加,提示右心室肥厚。上述结果表明,由PASMCs异常增殖导致的肺血管重塑在PAH的发病机制中占有重要地位。而目前大多数PAH的靶向治疗药物,其治疗靶点多为扩张肺血管,而对PASMCs的异常增殖无显著作用。因此,建立新的治疗思路,寻求新的治疗靶点,开发新的靶向治疗药物,抑制介导PASMCs增殖信号通路的关键环节,重建失衡的信号通路,预防甚至逆转PASMCs的增殖,应该是目前抑制肺血管重塑,治疗PAH的主要研究方向。
本研究显示二甲双胍通过激活AMPK抑制大鼠PAH模型的发生,这种保护作用伴随Skp2表达下调及P27蛋白水平上调,提示激活AMPK抑制野百合碱诱导大鼠PAH模型的保护作用可能与调控Skp2/P27信号通路有关。本研究为肺动脉高压的防治提供了新思路。
猜你喜欢
中国慈善家(2021年5期)2021-11-19 18:38:58
中国现代医药杂志(2020年12期)2020-02-06 06:32:16
心肺血管病杂志(2019年9期)2019-12-09 08:34:02
英语文摘(2019年6期)2019-09-18 01:49:08
乡村地理(2018年2期)2018-09-19 06:44:04
电子测试(2018年11期)2018-06-26 05:56:52
商周刊(2018年11期)2018-06-13 03:41:54
海峡姐妹(2018年4期)2018-05-19 02:12:44
民族音乐(2018年4期)2018-01-24 22:12:47
中国少年儿童(2017年16期)2017-07-18 11:15:11
