
Kdm2b基因敲入双loxp序列的NIH3T3细胞株的构建
2020-12-02 03:37陈素珠卢文显林典梁杜生荣林运鸿郑备红
山东医药 2020年32期
陈素珠,卢文显,林典梁,杜生荣,林运鸿,郑备红
1福建省妇幼保健院 福建医科大学附属医院,福州350001;2福建医科大学转化医学研究院
随着分子生物学技术的不断发展,基因敲除技术的应用越来越普遍。相应基因敲除的小鼠全基因组中都会出现基因表达缺失,常会影响其繁殖,无法对其发育后期的基因功能进行分析,这时候需要应用到条件性敲除小鼠。目前能实现条件性敲除的技术有Cre-loxp系统、FLPI系统等。Cre-loxp系统应用最普遍,由Cre重组酶和loxp位点组成[1~3]。借助基因编辑技术把两个loxp序列加入到目的基因的重要的外显子两端,制备出loxp小鼠即为条件性敲除小鼠,该目的基因表达不受loxp序列影响,当该小鼠与组织特异性表达cre重组酶的小鼠杂交后,cre重组酶可以使两侧loxp位点发生重组,目的基因完整性被破坏从而发生基因沉默。组蛋白去甲基化酶Kdm2b蛋白是组蛋白去甲基化酶家族JMJC蛋白的成员,在细胞周期、细胞凋亡、细胞衰老、肿瘤发生的调控中起重要作用[4]。Kdm2b敲除可致小鼠出现颅面部发育不全、脑瘫和低外显率的卷尾[5]。因此,条件性敲除小鼠为研究Kdm2b基因提供了载体。CRISPR/Cas9系统是一种新型基因编辑技术,通过sgRNA介导核酸酶Cas9对基因组特定位点进行识别、切割,从而实现基因编辑,操作相对简便,基因编辑效率高[6]。2018年7月~2019年2月,本研究利用CRISPR/Cas9系统和同源重组的原理将loxp序列插入到Kdm2b基因中,为后期制备条件性敲除鼠及基因功能研究进行初步探索。
1 材料与方法
1.1 主要实验材料 细胞、细菌和质粒:NIH3T3细胞(福建省发育与神经生物学重点实验室保存),pEASY-Blunt Cloning Vector、Trans1-T1(TransGen公司),PX459、PX458(Addgene公司),Vector 5(福建省发育与神经生物学重点实验室保存),Top10(TaKaRa公司),pBlueScript Ⅱ KS Vector(STRATAGENE公司)。试剂:Lipo2000(Invitrogen公司),嘌呤霉素(Life technologies公司),胎牛血清、青霉素/链霉素(Amresco公司),氨苄青霉素(上海生工公司),10×PCR Buffer、10×Loading Buffer(TaKaRa公司),RNase And DNase Away、质粒提取试剂盒(Roche公司),DMEM(Gibco公司),Wizard DNA Clean-Up核酸纯化试剂盒(Promega公司),T7E1(NEW ENGLAND Biolabs公司)。Kdm2b基因5臂正向引物序列为GTGCCCTCCTCTTACTCAGGTTTCG,反向引物序列为ATGTGGAAGTCAGTGAAACAGCCCT;Kdm2b基因3臂正向引物序列为GGCTACACCTTTTTCATCCCTTC,反向引物序列为ACCGACTATTACCTCCCTCATTG;Target正向引物序列为TCGAATTCACTCTGCAGTGGTAGGTCGCTCGCC,反向引物序列为TCGAATTCGCAGGAGGCTGCTTTCAAGTGCCGG;Donor DNA的5臂正向引物序列为CTGCAAGCCAAGGTGCTGTCTGTGA,反向引物序列为AGTCTCGAGCACCTGACTCAACAGCCT;Donor DNA的3臂正向引物序列为GCGTCGACAGTTCTGTGGTCTCAGCCAGGGCTG,反向引物序列为TAGAGGAAGACCCAAGGAAGACATT。主要仪器:电泳槽、凝胶电泳仪(北京六一公司),紫外凝胶成像系统(Syngene公司),PCR仪(5333型,Eppendorf公司),生物安全柜(HealForce公司),CO2恒温培养箱(Thermo公司),相差倒置显微镜(CKX-41型,Olympus公司)。
1.2 定点切割双链基因组DNA的sgRNA表达载体的构建 根据前期研究设计构建的sgRNA表达载体,在http://www.ncbi.nlm.nih.gov/网站上搜索小鼠Kdm2b基因序列,利用http://crispr.mit.edu/设计靶序列,在Kdm2b基因第7个外显子的5臂端筛选靶序列sgRNA5-2并在5臂加入BbsⅠ位点,sgRNA5正向序列为GGGAAACTCCACTTCGATAGAGG,PAM结构为AGG。同法在Kdm2b基因第9个外显子的3臂端筛选靶序列sgRNA3-2并在3臂加入BbsⅠ位点,sgRNA3正向序列为GAACCATTGACGTGCCCTGAGGG,PAM结构为AGG。根据所设计的序列合成sgRNA核苷酸链,退火后连接到已用BbsⅠ酶切后的线性pSpCas9(BB)-2A-Puro(PX459)质粒中,连接产物转化Transgenic1-T1感受态细胞中,挑取阳性单克隆菌落进行PCR鉴定,根据琼脂糖凝胶电泳结果判断sgRNA是否正确插入到载体中。
1.3 带有loxp序列的双链Donor DNA的设计与构建 分别根据前期筛选的sgRNA5和sgRNA3的切割位点设计对应的Donor DNA。Donor5 DNA作为sgRNA5同源重组的外源模板DNA,其3臂端具有EcoRⅠ酶切识别序列。Donor3 DNA作为sgRNA3同源重组的外源模板DNA,其5臂端具有EcoRⅠ酶切识别序列。分别用Donor DNA的5臂引物和Donor DNA的3臂引物从NIH3T3细胞基因组中克隆Kdm2b基因的5臂与3臂sgRNA位点的两侧同源臂片段。将5臂和3臂的同源臂片段插入带有loxp序列的Vector 5质粒中,提取质粒测序鉴定。根据loxp两侧的同源臂设计引物,以此前构建的带有同源臂片段的Vector 5载体作为模板,扩增出含有loxp序列(带EcoRⅠ酶切位点)的双链Donor DNA,测序鉴定。Kdm2b基因的Donor DNA设计情况见图1。

注:Donor5插入位点位于Kdm2b基因第7个外显子5臂端,与sgRNA5的切割位点相距50 bp;Donor3插入位点位于Kdm2b基因的第9个外显子3臂端,与sgRNA3的切割位点相距60 bp。
1.4 特定切割位点loxp序列敲入
1.4.1 sgRNA5切割位点的loxp序列敲入 利用脂质体转染法将构建好的Donor5和sgRNA5共同转染到NIH3T3细胞中,24 h后进行嘌呤霉素筛选8 d,形成阳性单克隆细胞。挑取阳性单克隆细胞,提取基因组。设计鉴定引物(5-鉴定引物-F&T-R)对基因组进行PCR扩增Kdm2b基因片段,获得产物,经过EcoRⅠ酶切后得到酶切产物,进行琼脂糖凝胶电泳并测序。
1.4.2 sgRNA3切割位点的loxp序列敲入 将已经在sgRNA5切割位点插入loxp序列的NIH3T3细胞进行培养,利用脂质体转染法将Donor3和sgRNA3共同转染到NIH3T3细胞中,24 h后进行嘌呤霉素筛选12 d,形成阳性单克隆细胞。挑取阳性单克隆细胞,提取基因组。设计鉴定引物(3-鉴定引物-F&3-鉴定引物-R)对基因组进行PCR扩增Kdm2b基因片段,获得产物,经过EcoRⅠ酶切后得到酶切产物,进行琼脂糖凝胶电泳并测序。
2 结果
琼脂糖凝胶电泳结果显示,sgRNA5和sgRNA3的PCR产物片段大小约200 bp,同时测序结果也正确,说明sgRNA5和sgRNA3都正确连接到PX459质粒上。Donor3和Donor5 DNA测序结果与设计相符合。将Donor5和sgRNA5共同转染到NIH3T3细胞中,选取阳性单克隆(图2),提取基因引物扩增、酶切鉴定,分别得到大小为2 000、200、350 bp的酶切产物,符合设计;该克隆基因组测序报告显示,loxp-EcoRⅠ序列正确插入到基因组中。见图3。将Donor3和sgRNA3共同转染到已插入一个loxp序列的NIH3T3细胞中,选取阳性单克隆(图4),提取基因引物扩增、酶切鉴定,分别得到大小为800、100 bp的酶切产物,符合设计;该克隆基因组测序报告显示,EcoRⅠ-loxp序列正确插入到基因组中。见图5。通过两次转染将loxp序列成功插入到Kdm2b基因第7个外显子和第9个外显子两端。

注:a、d为转染Donor5的NIH3T3细胞;b、e为转染Donor5和PX459的NIH3T3细胞;c、f为转染Donor5和PX459-sgRNA5的NIH3T3细胞。
3 讨论
KDM2B是JMJC家族的一个去甲基化酶,能特异性催化组蛋白H3第4位三甲基赖氨酸及组蛋白H3第36位三甲基赖氨酸去甲基化[7]。研究表明,KDM2B的作用包括抑制细胞衰老、分化、凋亡,维持细胞干性,促进细胞增殖、迁移和代谢等[8,9]。关于KDM2B在恶性肿瘤中作用的研究较多,但其在不同肿瘤中的作用各不相同,甚至可能出现相反的情况。在胰腺癌、胃癌、膀胱癌、鼻咽癌、卵巢癌中,KDM2B高表达会促进细胞增殖,使细胞更具侵袭性[10~13]。有学者发现,通过敲低小鼠胚胎成纤维细胞中KDM2B的表达可以抑制细胞增殖并诱导凋亡[14]。Konuma等[15]报道,在造血干细胞中KDM2B能与多梳蛋白形成复合物,以维持干性。同样的,顶端乳头干细胞中KDM2B过表达也会抑制细胞的分化[16]。Hong等[17]研究发现,KDM2B能与糖酵解相关基因Myc启动子区直接结合,抑制糖酵解并促进氧化磷酸化,抑制胃癌细胞增殖。另外,KDM2B还可通过MoMuLV来影响急性淋巴细胞白血病和急性髓细胞白血病的发生,当KDM2B过表达时诱导这两种白血病的发病,敲除KDM2B后可抑制细胞增殖并降低肺转移的发生率。然而,在HeLa细胞、人胶质母细胞瘤中过表达KDM2B可干扰这两种肿瘤细胞的增殖和代谢[18,19]。因此,在不同组织或类型的肿瘤中,KDM2B的生物学功能各不相同,其作用机制还不明确,有待进一步研究。
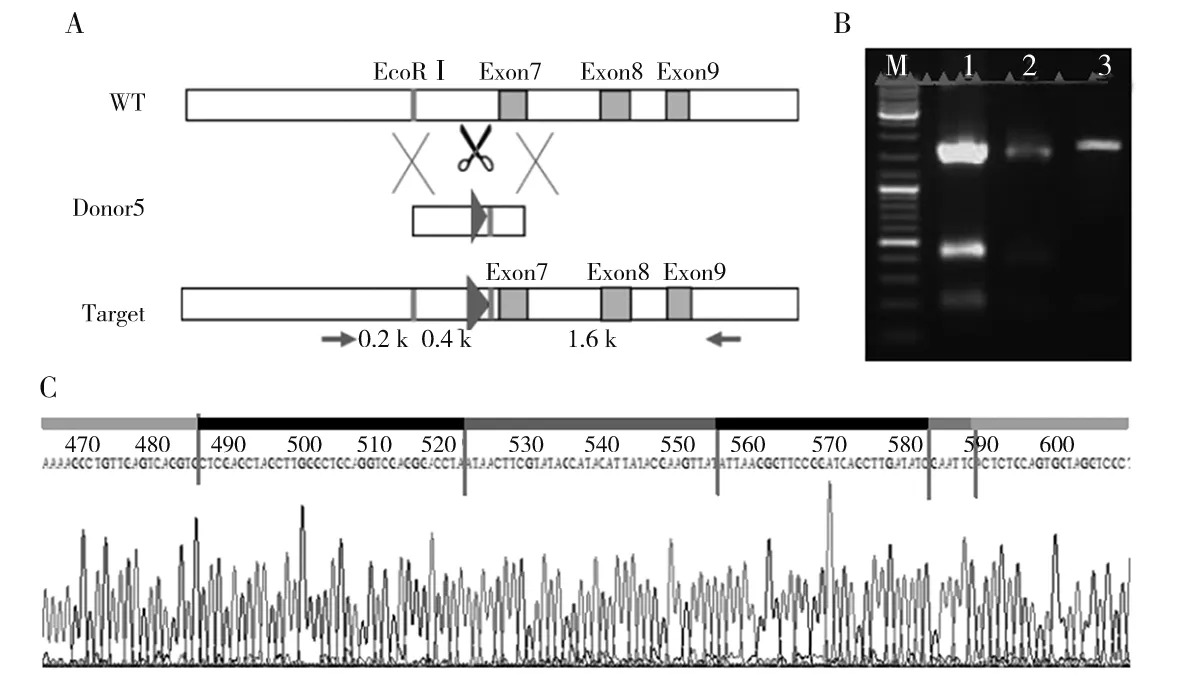
注:A为Kdm2b基因的5臂sgRNA的设计情况,其中WT为未经过基因编辑的Kdm2b基因组,外显子7上游有1个EcoRⅠ酶切位点,此位点下游为Donor5插入位置,Donor5带有的loxp序列下游也有1个EcoRⅠ酶切位点;Target为基因组中插入Donor5的Kdm2b基因,经酶切后可形成大小为2 000、200、350 bp的片段。B为转染Donor5和sgRNA5的NIH3T3细胞Kdm2b基因扩增产物电泳图,其中M为DNA Marker,1为阳性质粒,2为转染Donor5和PX459-sgRNA5的NIH3T3细胞,3为转染Donor5和PX459的NIH3T3细胞。C为转染Donor5和sgRNA5的NIH3T3细胞Kdm2b基因测序分析结果。

注:a、d为未经脂质体转染的NIH3T3细胞;b、e为转染Donor3和PX459-sgRNA3的NIH3T3细胞;c、f为转染Donor3和PX459-sgRNA3的NIH3T3细胞。
KDM2B是小鼠发育过程中重要的基因。利用动物模型研究KDM2B基因时通常需要敲除该基因。但是敲除KDM2B后,小鼠常常出现器官发育不全、生殖能力下降等问题。此时,需要通过条件性敲除的方式来避免这样的问题出现。Cre-loxp系统是最常用的一种条件性基因敲除系统。Cre-loxp系统其由Cre重组酶和loxp位点组成,是一种位点特异性重组系统[1]。loxp位点是一段由两个13 bp的反向重复序列和一个8 bp的不对称间隔区组成的DNA序列[2]。Cre重组酶可以特异性识别loxp位点,并引起两个loxp位点间的DNA序列发生特异性的重组[1~3]。Cre重组酶根据loxp序列在基因中的位置、方向不同可以敲除、倒转和染色体易位。当两个loxp序列在同一条DNA链且方向相同时,cre重组酶能敲除两个loxp序列直接的DNA片段[20]。通在条件性基因敲除小鼠的构建过程中,必须把两个loxp序列准确插入到需要敲除的基因的两侧。CRISPR/Cas9系统是利用sgRNA引导Cas9蛋白对特定基因位置进行基因切割的新型基因编辑技术。本研究采用同源重组的方式将loxp序列加入到特定位点,但是同源重组的发生率很低。通过引入CRISPR/Cas9系统,使同源重组位点DNA链发生断裂,修复的过程中发生同源重组的概率就会显著提高。本研究利用CRISPR/Cas9系统和同源重组的原理,通过两次转染将loxp序列成功插入到Kdm2b基因第7个外显子和第9个外显子两端,成功构建Kdm2b基因敲入双loxp序列的NIH3T3细胞株,为后期Kdm2b基因条件性敲除小鼠的制备和该基因的功能研究奠定了技术基础。
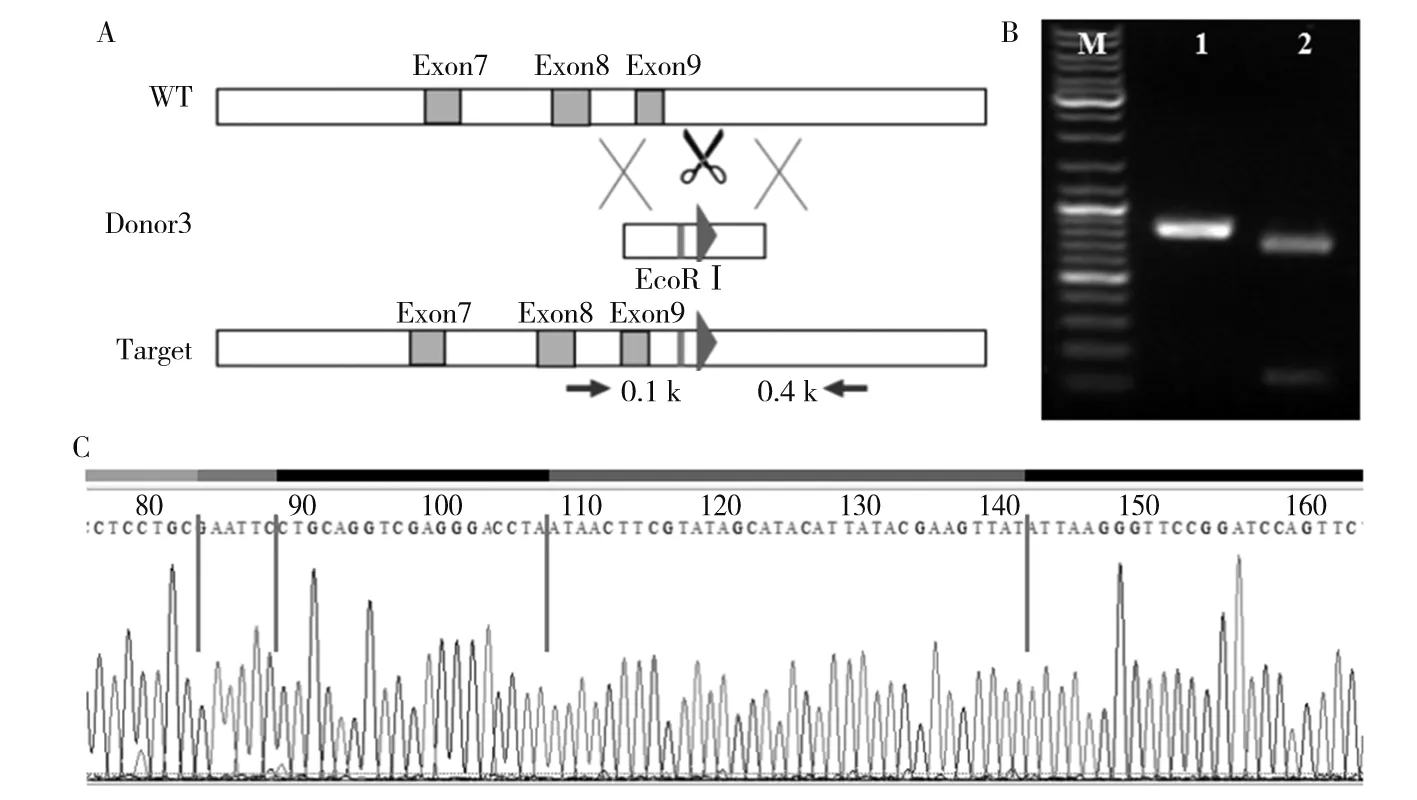
注:A为Kdm2b基因的3臂sgRNA的设计情况,其中WT为未经过基因编辑的Kdm2b基因组,外显子9下游为Donor3插入位置,Donor3带有的loxp序列上游也有1个EcoRⅠ酶切位点;Target为基因组中插入Donor3的Kdm2b基因,经酶切后可形成大小为100、700 bp的片段。B为转染Donor3和sgRNA3的NIH3T3细胞Kdm2b基因扩增产物电泳图,其中M为DNA Marker,1为阳性质粒,2为转染Donor3和PX459-sgRNA3的NIH3T3细胞,3为转染Donor3和PX459的NIH3T3细胞。C为转染Donor3和sgRNA3的NIH3T3细胞Kdm2b基因测序分析结果。
猜你喜欢
电子科技大学学报(2022年5期)2022-10-29
军事文摘(2022年16期)2022-08-24
基层中医药(2022年4期)2022-07-22
南京医科大学学报(自然科学版)(2021年8期)2021-10-19
汉字汉语研究(2021年2期)2021-08-30
今日农业(2021年11期)2021-08-13
中国生殖健康(2020年4期)2021-01-18
中国生殖健康(2020年4期)2020-12-09
中西医结合肝病杂志(2020年2期)2020-10-27
汉字汉语研究(2019年2期)2019-08-27
