
ETFDH基因杂合突变的脂质沉积性肌病1例
2020-11-30 04:35:22刘荣荣刘功禄马黎
温州医科大学学报 2020年11期
刘荣荣,刘功禄,马黎
(1.绍兴第二医院 神经内科,浙江 绍兴 312000;2.浙江大学医学院附属第二医院 神经内科,浙江 杭州 310000)
脂质沉积性肌病(lipid storage myopathy,LSM)是由于脂肪代谢过程中多种酶的功能缺陷或肉碱缺乏,影响了肌纤维内的脂肪酸代谢,导致异常增多的脂滴在肌纤维内堆积而引起一组疾病。笔者报告1例基因检测结果为ETFDH基因杂合突变的LSM,旨在提高对该病的认识与诊治水平,提高对该病分子遗传学的认识。
1 病例资料
1.1 现病史 患者,男,47岁,因“双下肢无力2年余”入院。患者2年余前无明显诱因下出现双下肢无力,行走时疲劳感明显,上楼梯时明显困难,蹲下起立困难,伴抬头无力,伴咬合力弱,无双上肢无力,无肢体麻木,2年来症状逐渐加重。于2019年2月18日入院。既往高血压病史2年,服药控制,血压控制不详。个人史:有吸烟习惯,吸卷烟,每天20支,已吸20年,未戒。既往史、婚育史、家族史无殊。
1.2 诊治过程 体格检查:内科系统无明显异常。神经系统:颈肌肌力 III级,胸锁乳突肌、咬肌肌力IV级,双侧三角肌、肱二头肌肌力V-级,对掌肌、分指肌肌力V级,髂腰肌肌力III级,股四头肌肌力IV级,胫前肌、腓肠肌肌力V-级,全身肌容积正常,四肢肌张力减低,感觉无异常,腹壁反射正常,双侧肱二、三头肌腱反射、桡骨膜反射(+),双侧膝腱反射、跟腱反射(+),双侧巴氏征(-)。生化检测示:肌酸激酶(creatine kinase,CK)为393 U/L,CK-MB 为37 U/L,乳酸脱氢酶为364 U/L,均高于正常值。肿瘤标志物检测:鳞状细胞癌抗原为1.62 ng/mL,高于正常值,其他血液学检查未见明显异常。大腿MRI:可见大腿后群肌(半腱肌、半膜肌、股二头肌)、臀大肌长T2、短T1,T2WI抑脂后信号减低(见图1)。肌电图示:肌源性损害与神经源性损害并存,神经源性损害表现为双下肢感觉波幅下降。基因检测:抽取患者外周静脉血,送广州欧蒙医学检验实验室行遗传性神经肌肉疾病相关基因检测;检验结果为阴性,附Sanger测序,ETFDH基因1395T>G杂合突变(见图2)。
结合以上病史、体格检查及辅助检查,诊断考虑肌病(疑代谢性肌病),建议上级医院就诊,完善肌肉病理等检查以进一步明确病情。
患者于2019年5月22日至2019年5月28日在浙江大学医学院附属第二医院神经内科住院治疗。入院诊断:肌无力待查。肌肉活检:取材左侧腓肠肌,光学显微镜下观察。肌肉病理结果见图3,符合LSM改变,明确诊断为LSM。给予维生素B2片20 mg tid口服治疗,1周后症状明显好转。1个月后随诊,患者基本恢复正常。
2 讨论
目前LSM的病因主要包括多种酰基辅酶A脱氢酶缺乏(multiple acyl-CoAdehydrogenase deficiency,MADD)、原发性肉碱缺乏、中性脂肪沉积症伴鱼鳞病和中性脂肪沉积症伴肌病[1]。MADD分为新生儿型和晚发型,我国最常见的LSM为晚发型MADD[2]。晚发型患者主要表现为肌肉无力和运动不耐受,肌肉病理多呈现典型的LSM改变。
MADD临床主要表现为青少年晚期或成年起病,临床表现异质性很大,表现为肌无力和运动不耐受,多有明显的颈肌无力和阻嚼肌受累。本病例以双下肢近端无力为主,伴有明显的颈肌和咀嚼肌无力。患者血生化示CK轻度或中度升高,发作期尿有机酸异常增多,血酰基肉碱分析显示中长链酰基肉碱明显增高。本例患者初次就诊时血生化提示CK轻度升高,以后的数次复查示CK轻度升高或正常,因条件限制,未做尿有机酸及血酰基肉碱检验。LSM患者肌肉MRI可见肌肉萎缩及脂肪沉积,研究表明晚发型MADD患者的大腿后肌群及臀肌的脂肪浸润最为显著,较大腿中间肌群和大腿前肌群脂肪浸润评分明显增高[3]。本例患者大腿肌肉MRI示臀大肌、大腿后群肌肉可见短T1,长T2信号,提示臀大肌及大腿后群肌脂肪沉积,T2WI抑脂后可见肌肉轻度水肿。神经电生理检查可见单独的神经源性损害、单独的肌源性损害、肌源性损害伴有神经源性损害,有学者认为周围神经受累的机制为脂质累及雪旺细 胞[4],并且经神经活检证实[5]。本例患者电生理示肌源性损害合并神经源性损害。病理染色见大量脂滴在肌纤维内沉积,以I型纤维为主。HE染色可见大量空泡样改变肌纤维,空泡在油红O染色被脂滴填充。遗传学方面,目前发现中国人群中的MADD导致的LSM均为ETFDH突变引起,69%为复合杂合突变,33%为纯合子突变,另有10%为单一杂合突变[2]。ETFDH基因编码电子转运黄素蛋白脱氢酶(ETF:QO蛋白),基因突变导致编码的ETF:QO蛋白异常,使得多种脱氢反应受损,导致肌纤维内积聚大量无法代谢的脂滴。
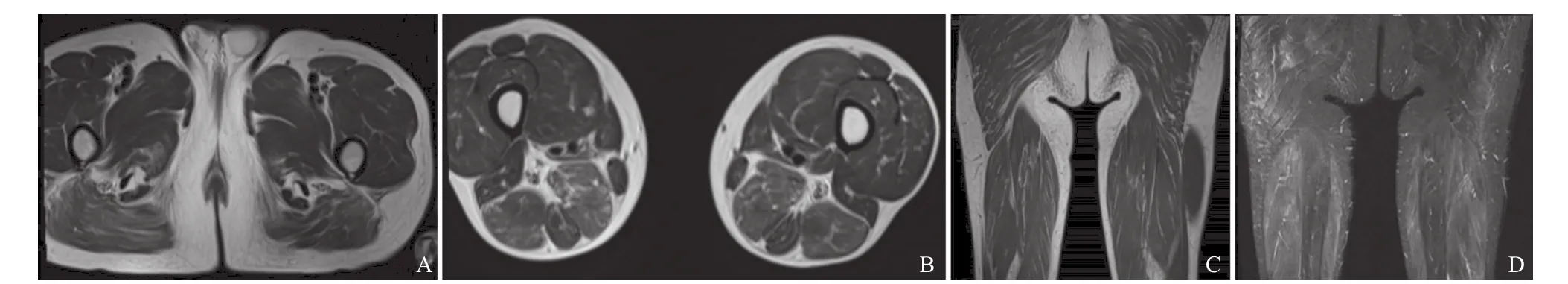
图1 LSM患者大腿MRI图像

图2 Sanger位点验证(ETFDH基因1395T>G杂合突变)

图3 肌肉病理结果
对于ETFDH突变引起的MADD,核黄素的治疗是有效的,由于核黄素为FAD的前体物质,当补充核黄素时可增加线粒体内FAD浓度,从而促进ETF:QO蛋白与FAD结合。单用核黄素治疗(30~120 mg/d),1~2周后临床症状开始改善,4~6周后肌力明显恢复,1~3个月后多数患者体力完全恢复正常[6]。
本病例ETFDH基因NM_004453.3:c.1395T>G(p.Y465*)为杂合突变,该基因为常染色体隐性遗传,通常杂合突变不致病。对于ETFDH基因突变导致的LSM,核黄素治疗可取得良好效果。由于该患者对核黄素治疗效果明显,考虑ETFDH基因复杂杂合突变为该患者的致病原因。笔者怀疑该基因另一条等位基因可能存在片段缺失或因为测序深度有限等原因,未发现另外一条等位基因的突变位点。c.1395T>G(p.Tyr465*)为无义突变,引起肽链合成提前终止。WANG等[7]报道1例MADD编号为53的患者仅检出c.1395T>G(p.Tyr465*)杂合变异,实验表明,该患者的ETFDH蛋白的含量显著降低。WEN等[8]对19例LSM患者研究中发现,17例患者考虑为MADD,其中有2例患者携带该位点的复合杂合变异。
综上,该病例在临床特点、生化、影像特点、神经电生理、病理、分子、治疗上均符合LSM最常见的分型晚发型MADD的特点,虽然基因检测结果为ETFDH基因杂合突变,我们考虑ETFDH基因复杂杂合突变可能为该患者的致病原因。
猜你喜欢
种子(2021年3期)2021-04-12 01:42:22
家庭百事通·健康一点通(2017年3期)2017-03-22 20:13:17
农村青少年科学探究(2016年5期)2016-11-29 11:24:56
外语教学理论与实践(2016年1期)2016-06-11 05:51:48
分析测试学报(2015年5期)2016-01-13 06:18:49
动物营养学报(2015年9期)2016-01-07 11:29:44
高中生学习·高一版(2014年6期)2014-07-05 10:20:16
华东理工大学学报(自然科学版)(2014年1期)2014-02-27 13:48:29
饲料工业(2013年6期)2013-04-08 03:51:17
山西农业大学学报(自然科学版)(2011年3期)2011-04-25 10:17:50
