
以抗血管生成为靶点的抗肿瘤药物研究进展
2020-11-27 03:57王钰艳宋明霞邓先清
聊城大学学报(自然科学版) 2020年1期
王钰艳 闫 瑞 宋明霞 邓先清
(井冈山大学 医学部,江西 吉安 343009)
0 前言
根据世界卫生组织(WHO)统计,全世界有3/5的人死于癌症、糖尿病、心血管疾病、慢性呼吸系统疾病这四大类疾病,而癌症则是最主要的死因之一[1].2018年的全球癌症负担状况报告估计到2018年,将有1810万新癌症病例和960万癌症死亡[2].虽然经过多年的发展,抗肿瘤药物的研发取得了许多重要进展.然而,面对威胁人类生命健康最严重的、占恶性肿瘤90%以上的实体瘤至今仍然缺乏高效、特异性强的药物,也反映了抗肿瘤药物研发的艰难.目前抗肿瘤药物研究呈现出多元化发展的趋势,其中以肿瘤血管生成作为靶点,开发血管新生抑制剂,逐渐成为抗肿瘤研究中一个重要的领域.
早在1971年,哈佛大学Folkman博士就提出了肿瘤的生长和转移具有血管依赖性的理论,并认为阻断肿瘤血管生成是有效策略[3].血管生成这个术语通常指新生毛细血管从原有的血管生成系统中萌发或分裂的过程,它在肿瘤生长的几个环节中起关键作用并有助于肿瘤的生长和促进转移[4].在血管生成过程中,内皮细胞获得独特的分子特征,并转化为血管生成表型,引起细胞行为和功能的各种变化.血管内皮生长因子(VEGFs)、表皮生长因子(EGFs)、血小板来源的内皮细胞生长因子(PDGFs)、酸性和碱性成纤维细胞生长因子是主要的血管生成介质以及较强的血管通透因子.最关键的因子血管内皮生长因子(VEGF)能促进血管通透性增加,并且血管内皮生长因子受体(VEGFR)的5个亚型中以VEGFR-2最重要.目前认为VEGF-VEGFR传导系统是肿瘤血管生成中主要的信号通路,VEGFR-2信号传导通路是生理性和病理性血管生成的关键.另外据文献报道,研究VEGFR-2抑制剂时应用最广泛的内皮细胞模型是人脐静脉内皮细胞(HUVEC)和鸡胚绒毛尿囊膜模型(CAM).
尽管由于抗血管生成药物广谱的抗肿瘤活性以及其相对较低的毒性,近年来发展较为迅速,但目前仍然存在一些问题.比如对晚期肿瘤患者疗效差、对不同肿瘤所用剂量存在较大的差异、长期使用易产生耐药性等[5],所以抗血管生成药物仍有很大的发展空间.本文通过查阅近年来靶向抗血管生成抗肿瘤药物的研究进展,对具有较好抗血管生成效果的抗肿瘤活性小分子进行了综述.基于靶点和骨架结构分类的方式对其发展状况、特性和前景进行了详细分析和总结,旨在为后续新的抗血管生成活性分子的发现以及结构优化工作提供思路.
1 以血管内皮生长因子受体- 2(VEGFR-2)为靶点的抗肿瘤药物
1.1 一些通过FDA批准的VEGFR-2抑制剂
如今,许多小分子VEGFR-2抑制剂已被合成并被批准为有效的抗癌药物,如Pazopanib、Sunitinib、Sorafenib、Axitinib、Cabozantinib和Foretinib等.临床经验表明,这些药物具有广谱抗肿瘤作用,不易产生耐药性.然而,导致治疗中断和剂量减少的副作用可能会抵消这些药物延长寿命的作用,这也表明对更安全的高效VEGFR-2抑制剂的需求没有得到满足.根据Mai Adel[6]所述,血管内皮生长因子受体(VEGFR-2)的几种小分子酪氨酸激酶抑制剂已经被FDA及临床批准用于治疗几种类型的癌症,它们的结构式及相应的抑制VEGFR-2的IC50值(见Figure 1).
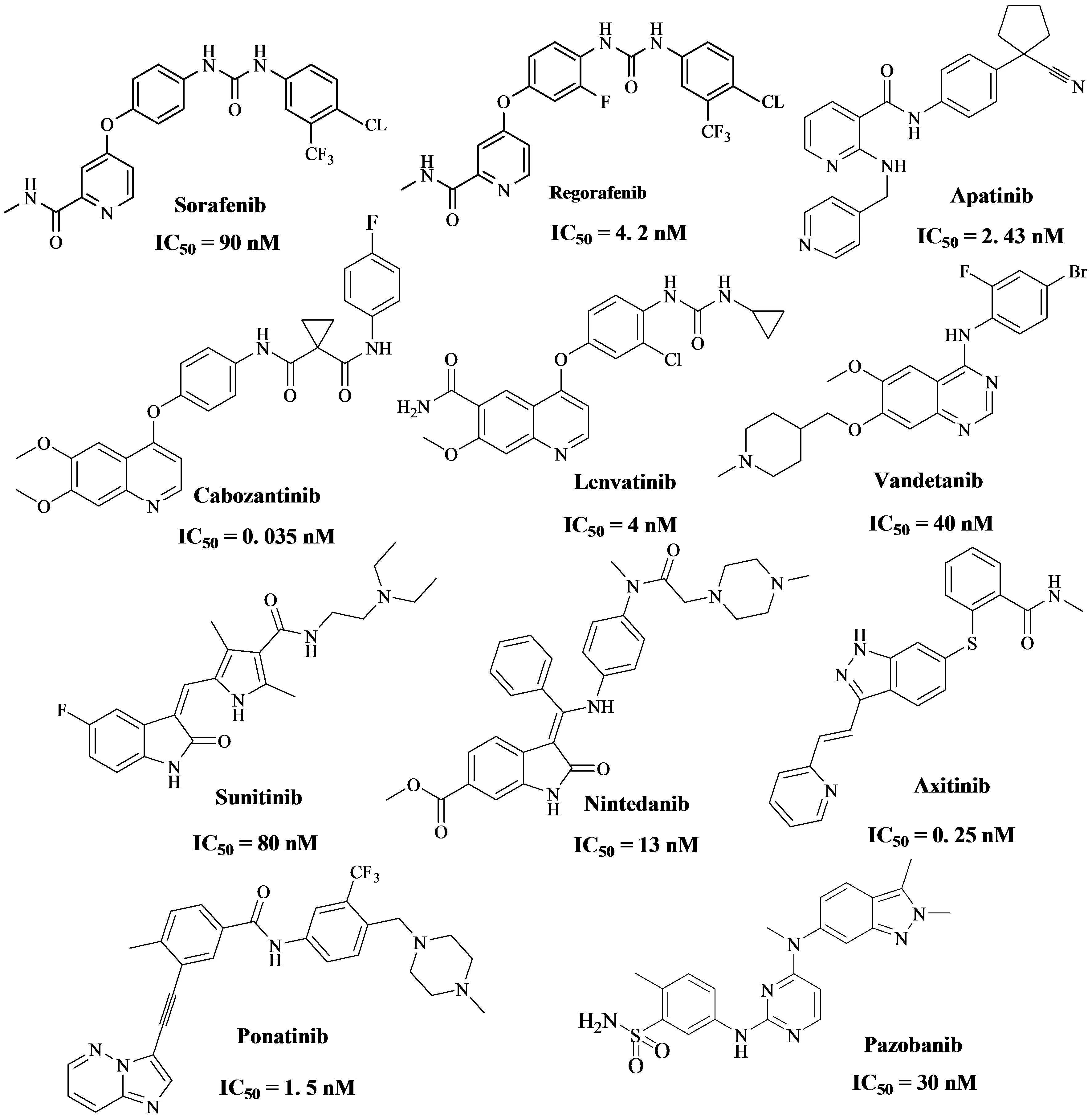
Figure 1 一些通过FDA批准的VEGFR-2抑制剂结构及相应IC50值
1.2 以嘧啶、噻吩环为基本骨架的VEGFR-2抑制剂
2018年,Mai Adel团队[6]以Sorafenib和2010年Yuya Oguro团队[7]合成的吡咯并[3,2-d]嘧啶衍生物(I)为比较对象,利用X射线共晶体结构进行结合模式分析后发现化合物(I)通过嘧啶环N与Cys919(半胱氨酸)的骨架-NH形成氢键,但Sorafenib与Cys919的羰基氧之间存在一个额外的氢键.Mai Adel团队合成的化合物不但可以保留基本氢键,而且重新排列吡咯-N的位置,使其可以与Cys919形成一个额外的氢键,从而达到模拟Sorafenib的目的,最终合成出了三个系列的新吡咯并[2,3-d]嘧啶衍生物即化合物1-3(见Figure 2),其中化合物1a和2a活性最强,抑制VEGFR-2激酶的IC50值分别为11.9 nM和13.7 nM.Amna Ghith团队[8]同样基于Sorafenib和化合物4,引入了吡喃和吡啶并[2,3-d]噻吩并嘧啶骨架,利用高极性基团来改善和增强药代动力学特性,从而设计出多种具有良好生物活性的配体.主要进行了两项结构修饰:第一次是用硫代嘌呤嘧啶骨架取代Sorafenib的吡啶环,用含氧同位素和氮同源衍生物取代非极性环己基.第二次修饰中,末端的芳香环被亲脂性基团(R)取代.最终合成出了三个系列噻吩并[2,3-d]嘧啶衍生物5-7(见Figure 2),化合物6a,7a和7b是最活跃的抑制剂,抑制VEGFR-2酶IC50值分别为2.5 μM,5.48 μM和2.27 μM.
经过对比分析,很显然Mai Adel团队合成的两个系列中化合物1a, 2a抑制VEGFR-2激酶的IC50值比Sorafenib(IC50=90 nM )更低,抑制效果很强,相比之下Amna Ghith团队合成的系列衍生物IC50值均处于μM级别,抑制效果没有前者突出,但是药代动力学特性上不一定前者比后者效果更好,可以进行后续的进一步研究来证明.
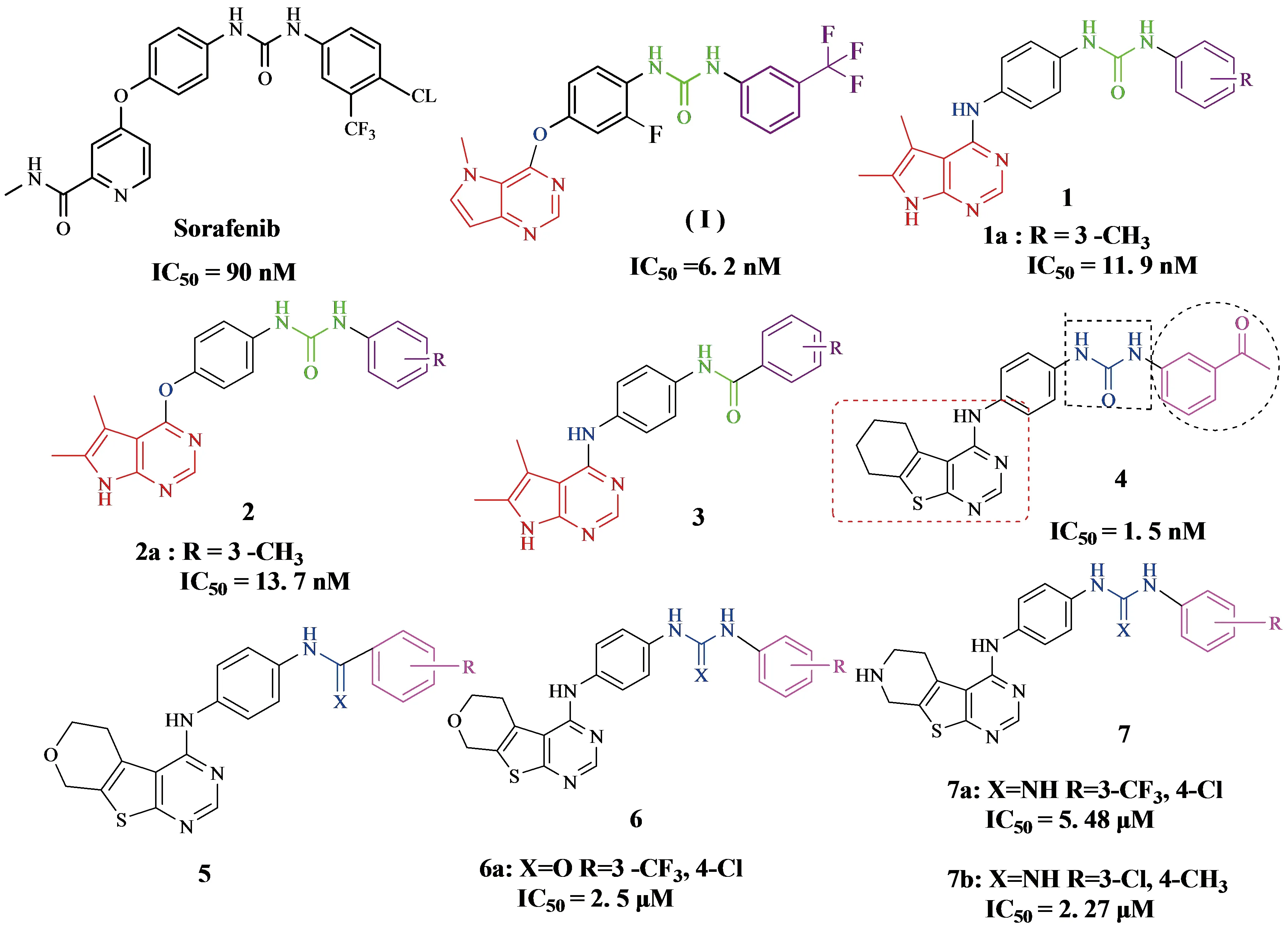
Figure 2 Mai Adel团队、Amna Ghith团队合成的系列衍生物
2016年,Hao Qiang团队[9]设计合成了一系列4-氨基嘧啶-5-甲醛肟类化合物, 他们认为c-Met与VEGFR-2具有协同作用,进而推测双重抑制c-Met和VEGFR-2的分子可能具有更广谱的优势.基于喹唑啉骨架是一种已经被证明的活性骨架,目前只报道为有效的VEGFR-2抑制剂[10],而4-氨基嘧啶-5-甲醛肟因其分子内氢键形成了一个伪六元环被认为是喹唑啉的等立体结构.Cabozantinib和Foretinib都是治疗甲状腺髓样癌的化合物,此前有报道称它们同时抑制c-Met和VEGFR-2[11].化合物8a (见Figure 3)与蛋白c-Met和VEGFR-2对接,可以作为进一步研究c-Met和VEGFR-2双抑制剂的先导分子.
2017年,Jieming Li团队[12]也设计合成了一系列噻吩[2,3-d]嘧啶衍生物作为新型双c-Met和VEGFR-2激酶抑制剂,将噻吩并[2,3-d]嘧啶骨架引入侧链基合成了一系列化合物9 (见Figure 3),其中化合物9a (见Figure 3) 抑制活性最强.两个团队的设计思想很相近,都插入了环丙烷-1,1-二羧基酰胺基结构,但是Jieming Li团队的化合物系列抑制效果更强.
2018年,Guoshun Luo团队[13]和Wuji Sun团队[14]也各自相继合成出了活性较好的嘧啶衍生物.Guoshun Luo团队着重于研究二取代嘧啶衍生物即化合物10-12 (见Figure 4)的构效关系,而Wuji Sun团队以Pazopanib、Sorafenib、Axitinib三种VEGFR-2抑制剂的结构片段为基础,将它们的活性骨架组合成了三个系列嘧啶衍生物即13-15 (见Figure 4).
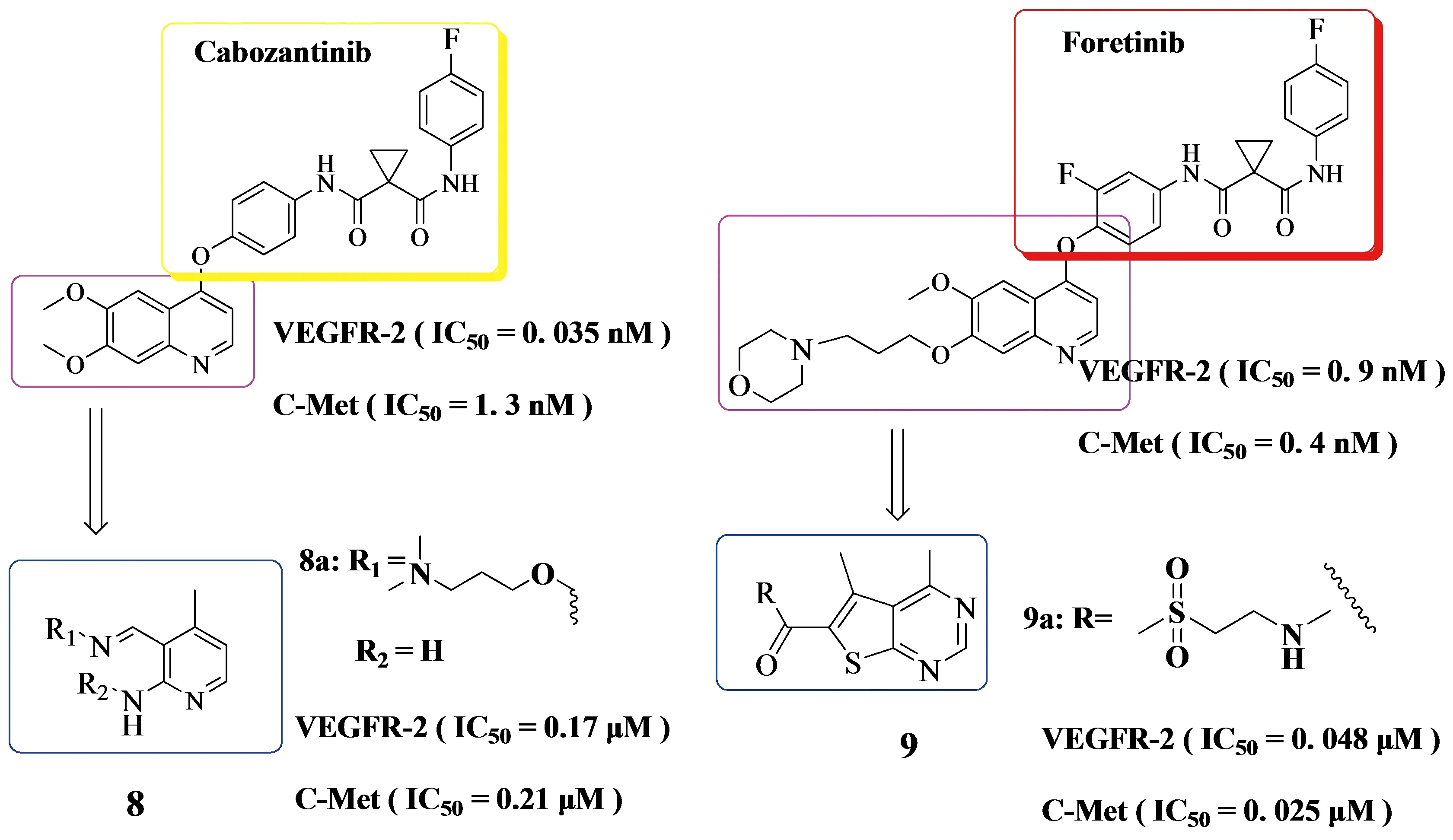
Figure 3 Hao Qiang团队、Jieming Li团队合成的系列衍生物
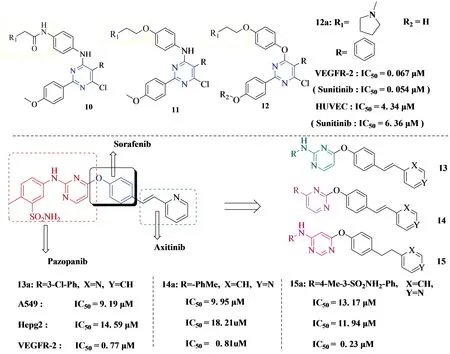
Figure 4 Guoshun Luo团队、Wuji Sun团队合成的系列衍生物
1.3 以喹啉、喹唑啉、吲哚啉为骨架的VEGFR-2抑制剂
2016年,Liang Lu团队[15]基于ZD4190和Tivozanib中的4-苯胺-喹唑啉和4-羟基喹啉活性骨架,将4-芳基羟基引入喹唑啉骨架中,得到了以4-羟基喹唑啉为核心骨架的一系列喹唑啉衍生物.并且他们还在4-羟基喹唑啉核的基础上合成了一系列基于席夫碱骨架的喹唑啉类似物16 (见Figure 5).其中化合物16a活性最好,HeLa ( IC50=0.22 μg/mL),A549 ( IC50=0.15 μg/mL ),MCF-7 ( IC50=0.24 μg/mL ).2018年,Prateek Pathak团队[16]也合成了一系列喹唑啉团簇1,3,5三嗪衍生物17 (见Figure 5).据报道,杂环1,3,5-三嗪及其衍生物是多种治疗靶点的有效骨架.单、二或三氨基取代的1,3,5-三嗪偶联物如三胺嗪和dioxadet已经被报道为抗癌药物[17].化合物17a,17b,17c活性突出,依次对HeLa(IC50=1.94,1.84,1.82 nM),MCF-7(IC50=1.9,1.84,1.85 nM),HL-60(IC50=1.32,1.25,1.29 nM),Hepg2(IC50=0.98,0.98,0.63 nM),并且与Vandatentinib药品(IC50=1.87,1.87,1.72,0.33 nM)对比分析,三个化合物对该四种细胞的活性均强于Vandatentinib.
2015年,Wagdy M.Eldehna团队[18]受到Sorafenib,Regorafenib,Sunitinib三个抗肿瘤药物结构的启发,合成了一系列以吲哚啉脲为结构的化合物18 (见Figure 5),其中伊沙汀(1H-吲哚-2,3-二酮)是最有前途的一类杂环化合物,具有多种有趣的活性特征,在人体中具有良好的耐受性[19].最终实验数据表明当R基为4-SO3NH2,X为Cl原子时活性最强.对VEGFR-2(IC50=0.31μM),Hepg2(IC50=3.15μM),而对照品Sorafenib对VEGFR-2(IC50=3.4 μM),Hepg2(IC50=0.1 μM),由此可见,当R基为4-SO3NH2,X为Cl时的化合物确实对VEGFR-2的抑制作用甚至比Sorafenib强.
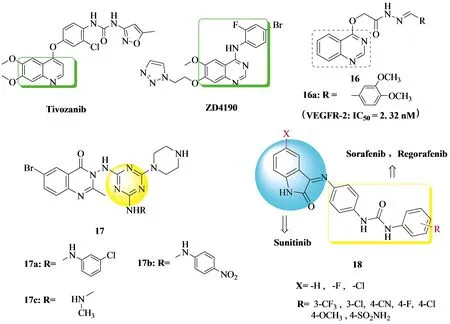
Figure 5 Liang Lu、Prateek Pathak、Wagdy M.Eldehna团队合成的系列衍生物
2 以成纤维细胞生长因子受体(FGFR)为靶点的抗肿瘤药物
成纤维细胞生长因子受体(FGFR1-4)是酪氨酸受体激酶(RTKs)的一个亚家族,参与许多细胞过程,如血管生成、胚胎生成、组织稳态、伤口修复和癌症等.大量证据表明,FGF/FGFR信号的激活在肿瘤的进展和生长中起着关键作用[20].此外,FGF/FGFR信号异常在多种癌症中频繁出现,使FGFR成为抗癌药物开发的热点靶点之一.
2.1 以吡咯并吡嗪、吡唑并吡啶环为基本骨架的FGFR抑制剂
2018年,Alan Jiang团队[21]合成了一系列5H-吡咯并[2,3-b]吡嗪FGFR激酶抑制剂.在这之前他们报道过几个化合物作为c-Met抑制剂的生物活性,以1-磺基吡唑啉[4,3-b]吡啶为骨架的化合物具有较好的活性[22].经过构象分析,发现其中化合物19的整体结合构象是反向的,这说明甲基吡唑指向FGFR1的ATP位点的后囊,与其它的有显著的差异.在接下来的工作中,将1H-吡唑并[4,3-b]吡啶骨架改为5H -吡咯并[2,3-b]吡啶从而合成了化合物20和21(见Figure 6)[23],发现可以增加与FGFR1结合的活性.不过最终他们选择5H-吡咯并[2,3-b]吡嗪作为进一步修饰的骨架,于是合成了一系列5H-吡咯并[2,3-b]吡嗪衍生物.活性最好的化合物22a与FGFR1结合的方式与化合物19结合的方式非常相似,并进一步对化合物22a的咪唑基团进行了修饰,并对其FGFR1抑制活性进行了评价,结果表明,以乙基和异丙基取代咪唑环,化合物间具有较好的抑制活性.
2016年,Bin Zhao团队[24]探索并合成了一系列取代的1H吡唑并[3,4b]吡啶衍生物作为有效的和选择性的FGFR激酶抑制剂.他们基于对AZD4547结构的探索,发现这个骨架对FGFR具有更强的抑制效果和选择性,并且利用骨架跳跃策略,在苯环的2位和6位掺入Cl,设计了新型1H-吡唑并[3,4-b]吡啶骨架衍生物23(见Figure 6),活性最好的化合物23a对FGFR1的IC50达到0.3 nM.
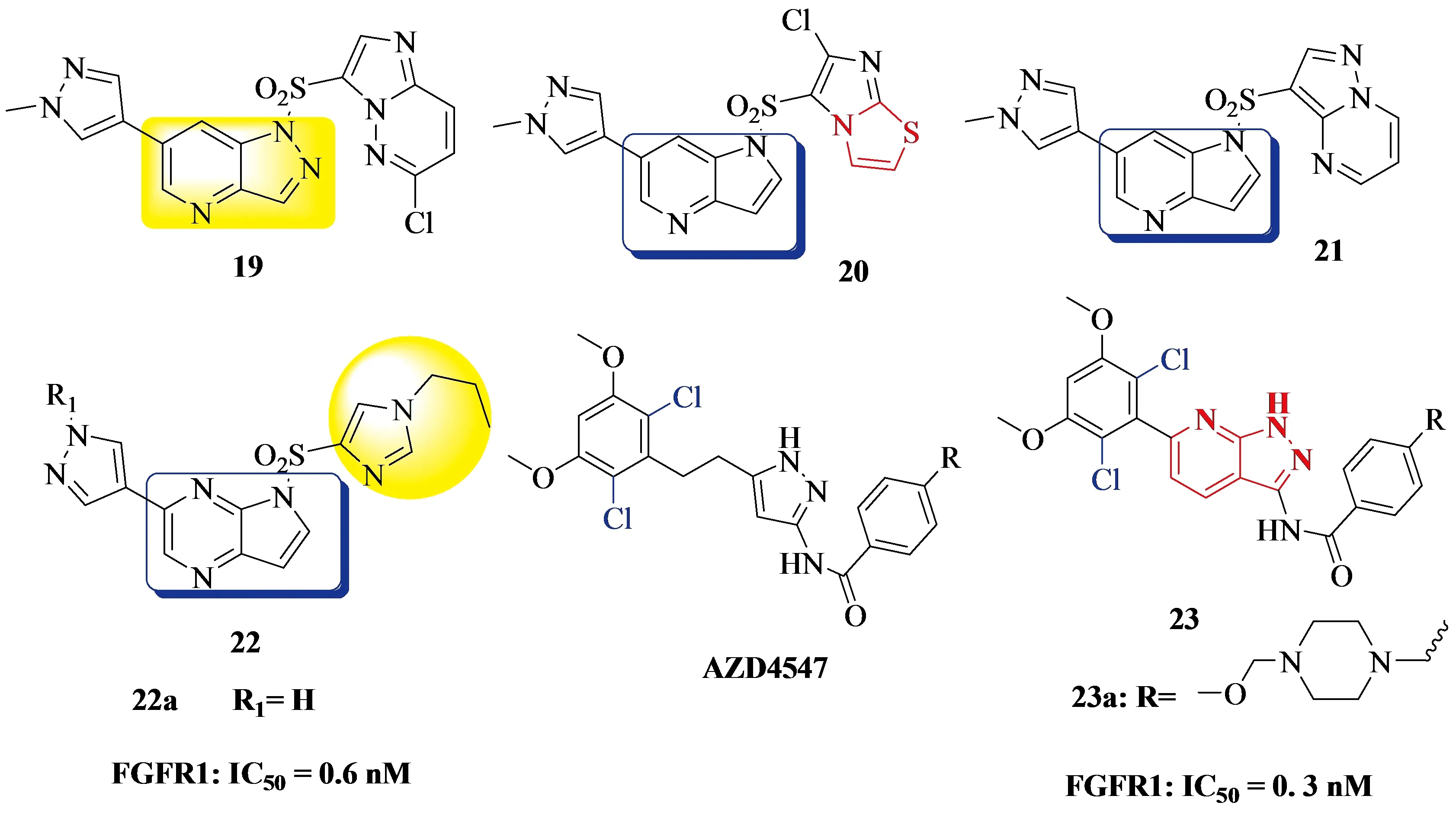
Figure 6 Alan Jiang团队、Bin Zhao团队合成的系列衍生物
2.2 以吲哚唑环为基本骨架的FGFR抑制剂
2015年,Jian Liu团队[25]就合成过一系列吲哚唑类FGFR抑制剂24 (见Figure 7).基于AZD4547和NVPBGJ-398的结构,NVP-BGJ398是由诺华公司开发的一种选择性FGFR抑制剂,具有良好的FGFR1和FGFR2抑制作用(FGFR1:IC50=0.9 nM;FGFR2:IC50=1.4 nM)[26],他们利用骨架跳变和分子杂交策略设计了28种新型1H-吲哚-3-胺骨架衍生物.含6-(3-甲氧基苯基)-1H-吲哚唑-3-胺骨架的化合物24a首次被
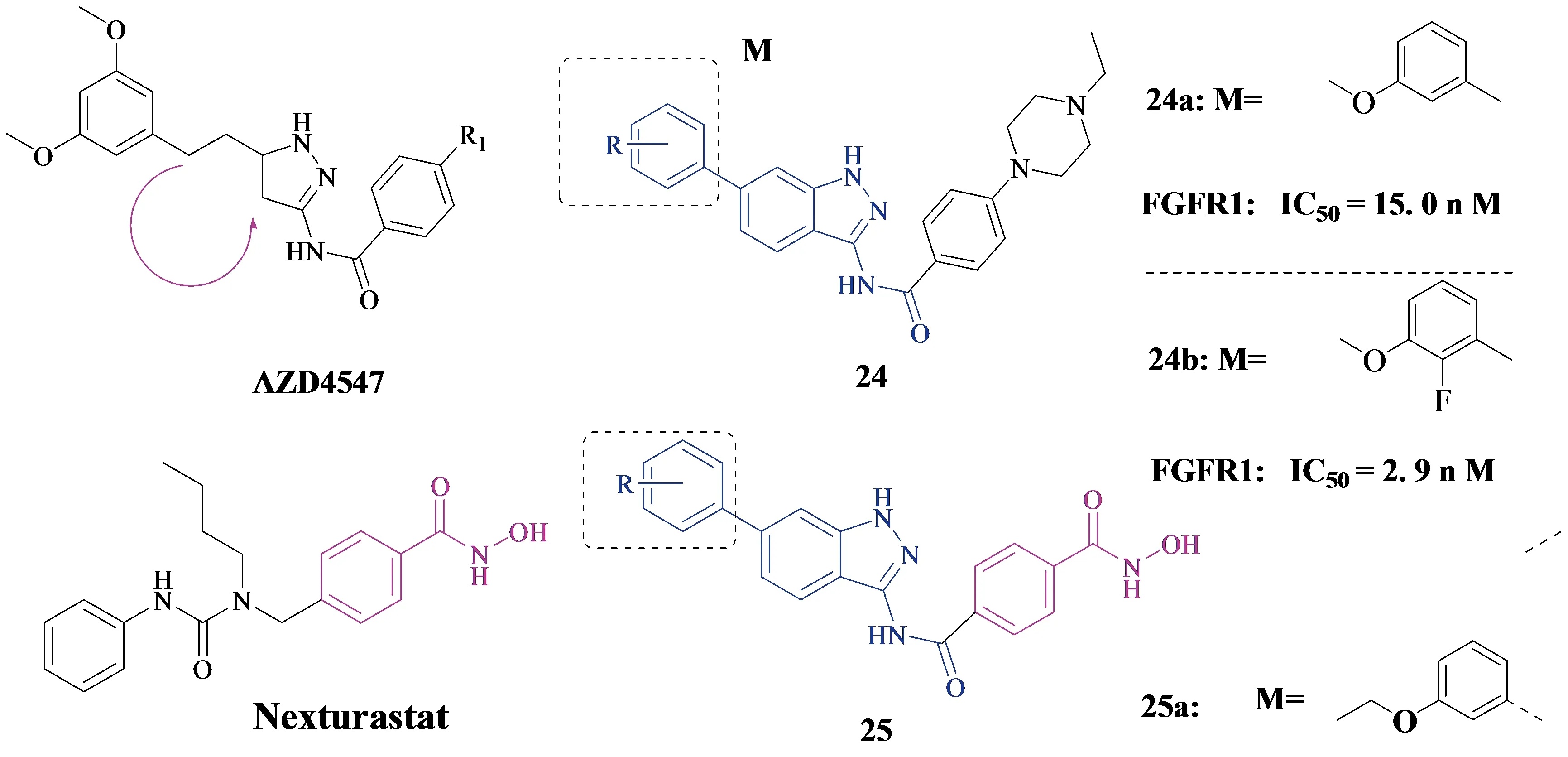
Figure 7 Jian Liu团队合成的吲哚唑类FGFR抑制剂
鉴定为一种有效的FGFR1抑制剂,具有良好的酶抑制作用.研究化合物24a与FGFR1结合的晶体结构,进一步的结构优化表明,化合物24b是最有效的FGFR1抑制剂,具有最佳的酶抑制作用和细胞活性(见Figure 7).
2017年,该团队[27]还继续合成了一系列吲哚唑类FGFR1和HDAC双重抑制剂25(见Figure 7).组蛋白去乙酰化酶(HDAC)也是肿瘤治疗的重要靶点.虽然双HDAC药物载体联合酪氨酸激酶抑制剂(TKIs)已取得成功,但双HDAC/FGFR1抑制剂尚未见报道.因此他们设计了一系列带有1H -吲哚唑-3-胺和苯并羟肟酸骨架的杂化产物.据报道,有许多关于苯并羟肟酸作为选择性HDAC6抑制剂,比如Nexturastat[28],其中化合物25a对HDAC6和MCF-7的抑制作用最强,IC50为34 nM和 9 μM.同时,该化合物还表现出一定程度的FGFR1抑制活性,为进一步探索双HDAC/FGFR1抑制提供了基础.
3 以表皮生长因子受体(EGFR)为靶点的抗肿瘤药物
表皮生长因子受体(EGFR)是一类跨膜生长因子受体PTKs.EGFR家族有四个成员:HER2(人表皮生长因子受体-2)及其近亲HER1,HER3和HER4.EGFR的表达与许多上皮性肿瘤(如结肠癌、乳腺癌、卵巢癌和NSC肺癌)的产生有关[29].它将通过与ATP竞争其目标酶催化域的结合而发挥作用.许多小分子EGFR激酶抑制剂已在癌症临床试验中得到评价.例如,已批准用于晚期非小肺癌化疗治疗的含苯胺喹唑啉化合物厄洛替尼和吉非替尼.
3.1 以喹唑啉、噻唑啉为基本骨架的EGFR抑制剂
EGFR和HER2在乳腺癌、卵巢癌、前列腺癌等多种肿瘤中均有表达,但是患者愈后效果差[30].因此,双靶点EGFR/HER2比单抑制EGFR更有效.因为EGFR/HER2抑制剂可阻断酪氨酸激酶磷酸化,抑制肿瘤细胞内信号上调,导致肿瘤调控功能丧失.已经有多种与ATP竞争的EGFR/HER2 RTK双抑制剂与不同骨架相关的报道,许多目前正在市场或临床试验中用于治疗癌症.
Ahmed Elkamhawy团队[31]于2017年就合成了一系列具有6-取代-4-苯胺喹唑啉核的新化合物26(见Figure 8).EGFR/HER2双抑制剂的一个重要先导核是4-苯胺喹唑啉,先导化合物拉帕替尼已被FDA批准用于治疗HER2过度表达转移性乳腺癌患者[32].尽管拉帕替尼已被证明是治疗有效的,但还是有许多患者治疗无效果或变得更耐药.新化合物在取代的4-苯胺喹唑啉的C-6上引入新的基团或改变C-4苯胺基团来提高效果和选择性.他们报道的化合物26a,26b,26c (见Figure 8)活性突出,对BT-474 细胞毒性依次为(IC50=2.70,1.82,1.95 μM),抑制EGFR和HER2的IC50值依次有27a ( IC50=0.003,0.016 μM ),27b ( IC50=0.035,0.126 μM),27c ( IC50=0.227,1.390 μM ).
同一年,Yu-Jia Ren团队[33]也合成了含有吡啶、吡唑和噻唑啉酮的新型抗癌衍生物(见Figure 8).近年来,噻唑啉酮类化合物以其抗肿瘤活性成为新的研究热点,如包含苯并噻唑结构的噻唑啉酮其抗癌活性已被证明[34].许多吡啶、吡唑或噻唑啉酮的衍生物也被报道过有强大的生物活性以及低毒性[35].化合物26a的活性最为突出,抑制EGFR ( IC50=0.099 μM ),HER2 ( IC50=3.26 μM ),对B16-F10,HeLa和MCF-7三种细胞的抑制活性依次为(IC50=0.09 μM,0.29 μM,0.56 μM ).
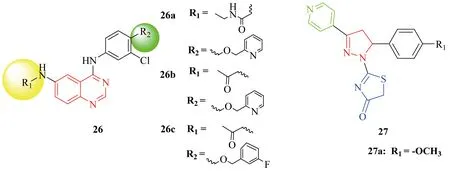
Figure 8 Ahmed Elkamhawy团队、Yu-Jia Ren团队合成的EGFR抑制剂值
3.2 以吡唑、吡嗪环为基本骨架的EGFR抑制剂
2015年,Mohamed A团队[36]合成了一系列新型吡唑并[3,4-d]嘧啶衍生物EGFR-TK抑制剂28(见Figure 9).该吡唑并[3,4-d]嘧啶衍生物被认为是嘌呤核的同分异构体,具有良好的抗肿瘤活性,因此受到广泛关注,而希夫碱也被发现是一种很有前途的新型抗癌药物骨架[37].该团队对含苯胺喹唑啉化合物的一般特征进行了化学修饰.这些修饰包括用吡唑基取代喹唑啉骨架中的苯环部分,以增强细胞毒性.另一种修饰是在吡唑啉嘧啶核的C-4上引入席夫碱,目的是利用基于碎片的药物设计方法来开发更有效的靶向分子.实验结果显示,化合物28c、28d、28g活性最强,三个化合物对EFGR(表皮生长因子受体)的抑制活性(见Figure 9).对MCF-7细胞的抑制活性依次为(IC50=7.22 μM、8.74 μM、6.14 μM ); 对A549细胞的抑制活性依次为(IC50=13.59 μM、14.42 μM、1.17 μM ); 对HT-29细胞的抑制活性则依次为(IC50=8.20 μM、7.35 μM、5.36 μM ).
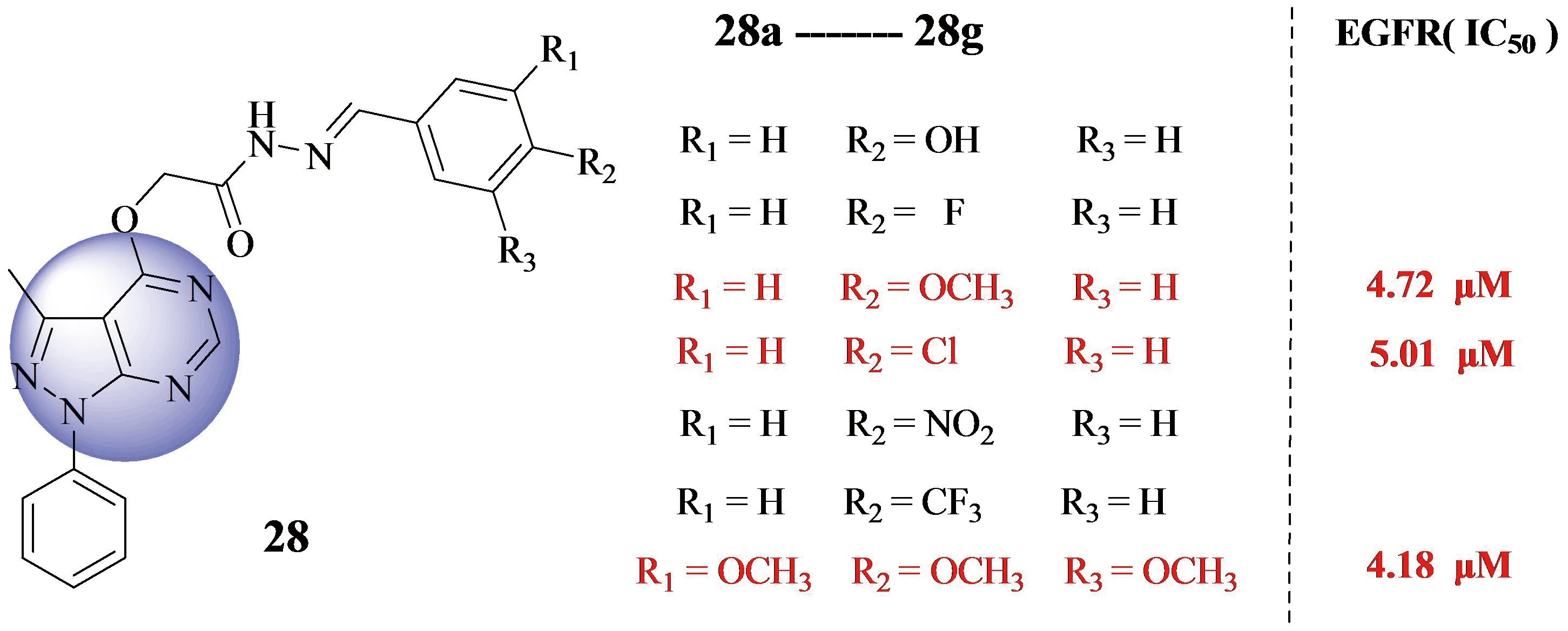
Figure 9 Mohamed A团队合成的新型吡唑并[3,4-d]嘧啶类EGFR抑制剂
4 以蛋白激酶(AKt)为靶点
Akt(蛋白激酶B)是PI3K/Akt/mTOR信号级联的关键连接,调控细胞生长、增殖、存活和凋亡.磷脂酰肌醇-3激酶(PI3K) / Akt通路是人类癌症中最常见的信号通路之一.Akt是这一通路的关键成分,在多种人类癌症中过度表达或激活,包括胶质瘤、肺癌、乳腺癌等.抑制Akt信号通路能使Akt的活性升高从而导致肿瘤细胞凋亡.因此,Akt已被证实是一种可行的抗癌药物靶点,目前有几种有前景的Akt抑制剂正处于不同的临床评估阶段[38].
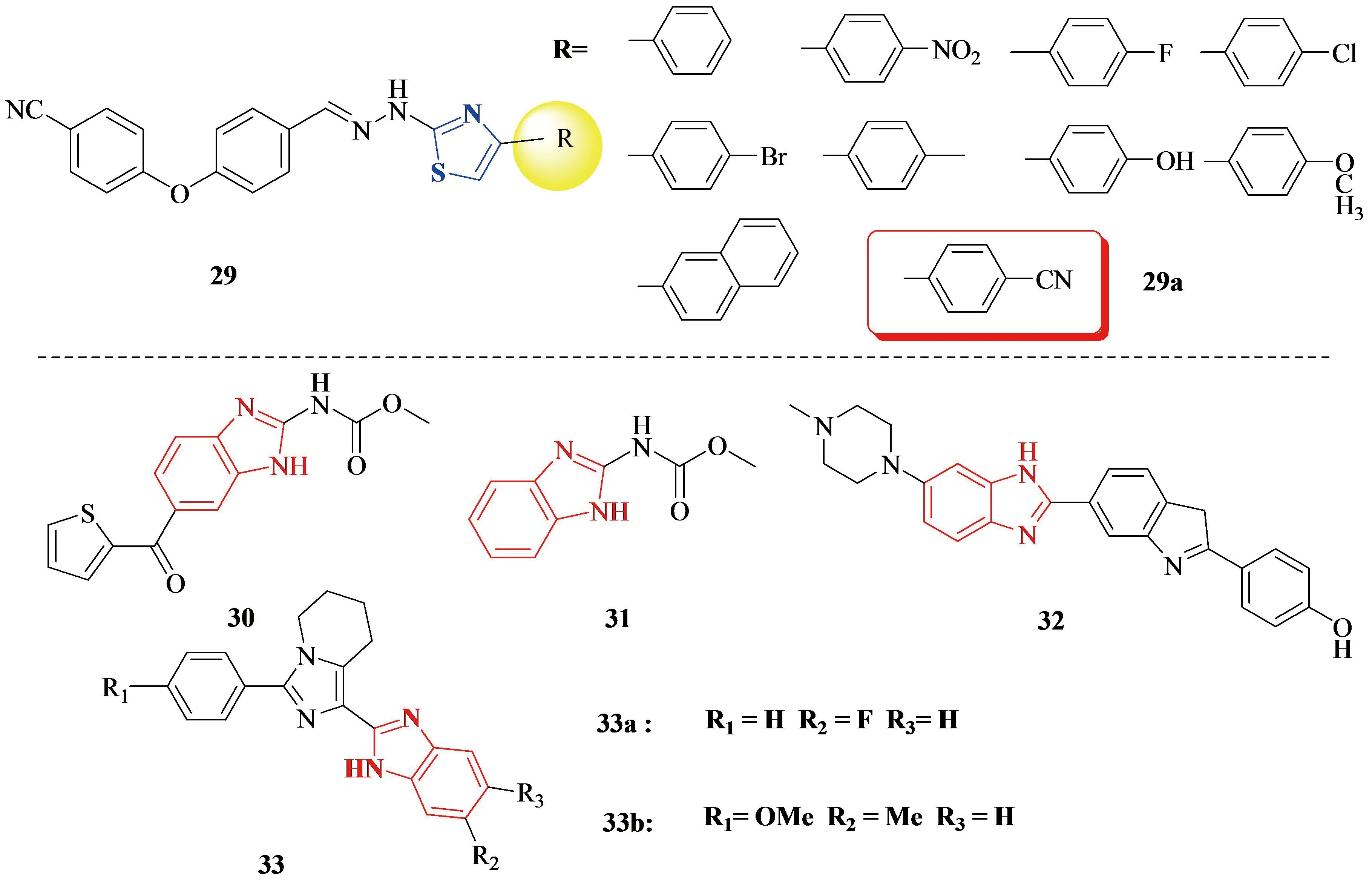
Figure 10 Mehlika Dilek Altinto、Ahmed Kamal团队合成的噻唑及苯并咪唑类Akt抑制剂
4.1 噻唑、咪唑类AKt抑制剂
2018年,Mehlika Dilek Altinto团队[39]为了开发针对Akt的高效抗癌药物,合成了新的噻唑衍生物29(见Figure 10).噻唑作为一种多用途骨架材料,因其在先导物鉴定和优化方面的重要作用而受到广泛关注.含有多种官能团的异取代噻唑衍生物广泛存在于许多天然化合物中,如硫胺素和商业合成药物中.最有希望的抗癌药物化合物29a(见Figure 10),其选择性抑制A549和C6细胞的IC50值分别为12.0 μg/mL和3.83 μg/mL.
2014年早期,Ahmed Kamal团队[40]也合成过一系列咪唑-[1,5-a]吡啶-苯并咪唑衍生物33(见Figure 10)作为PI3K/Akt通路的抑制剂.含咪唑环的氮-桥头融合杂环是具有重要药理作用的分子中常见的结构,对多种靶点具有广泛的活性.应用最广泛的杂环系统之一咪唑吡啶类化合物,具有广泛的生物活性如芳香化酶抑制剂、雌激素生成抑制剂、血小板聚集抑制剂等.咪唑吡啶通过抑制血管内皮生长因子受体和诱导细胞凋亡等不同的分子机制表现出抗癌活性.NSC-238189(30)、FB642(31)和Hoechst-33258(32)等化合物[41]都是具有苯并咪唑结构的抗肿瘤药物.实验结果表明化合物33a和33b显示明显的细胞毒性其GI50值分别从1.06 - 14.9 μM和0.43 - 7.73 μM.对乳腺癌细胞的IC50值分别为1.79 μM,2.85 μM.流式细胞术分析表明,这些化合物在G2/M期阻滞细胞周期,并通过凋亡机制诱导细胞死亡.
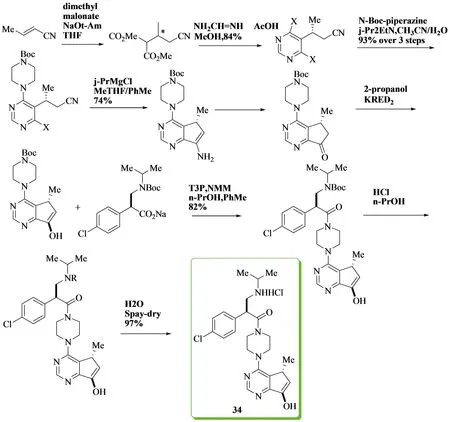
Figure 11 Chong Han团队进行不对称合成Ipatasertib最佳反应工艺路线
4.2 第一代Akt抑制剂:临床期药物Ipatasertib的合成
Chong Han团队[42]于2017年进行不对称合成的药物GDC-0068(Ipatasertib)是一种新型有效的抑制剂,可以靶向所有Akt激酶三种亚型的ATP结合裂解.Ipatasertib即化合物34(见Figure 11)目前正在进行III期临床试验,用于治疗转移性去势性前列腺癌和三阴性转移性乳腺癌.活性药物成分(API)是一个复杂的6,7-二氢-5H-环戊烷[d]嘧啶哌嗪酰胺类化合物,包含三个手性中心的组装手性双环嘧啶和手性α-芳基-β-氨基酸.为了进一步挑战工艺化学,该团队选择了具有高度吸湿性和潮解性的原料药单盐类化合物进行开发.通过摸索众多反应条件其最佳反应工艺(见Figure 11)
5 以局灶性粘附激酶 (FAK)为靶点
局灶性粘连激酶(FAK)是一种定位于局灶性粘连的细胞质酪氨酸激酶和骨架蛋白,同时也是肿瘤微环境中细胞信号传递的重要功能调控因子.这种蛋白质正在成为一个有前途的治疗目标,因为它在转录和翻译水平过度表达于各种癌症,包括胰腺癌、卵巢癌、宫颈癌、前列腺肿瘤、头颈部鳞状细胞癌、胶质母细胞瘤、结肠癌、乳腺癌、肺癌和肾癌等.并且大量证据表明,FAK信号通路通过调控细胞迁移、侵袭和血管生成,可以调控肿瘤的进展和转移形成[43].
2013年早期,Pascal Dao团队[44]先合成了一系列1,3,5-三嗪类局部粘附激酶抑制剂35 (见Figure 12),并且最近被证明对HUVEC细胞具有抗血管生成活性,对多种癌细胞具有抗癌作用.他们研究了诺华制药公司设计的TAE-226对HUVEC细胞的活性,发现最有效的化合物35a (见Figure 12) 的共晶结构的x射线结晶学分析表明,该化合物与FAK激酶结构域的相互作用模式与在TAE-226络合物中观察到的非常相似.
之后2017年,Pascal Dao团队[45]再次设计并合成了一系列含1,2,4-三嗪核的新化合物36 (见Figure 12) 作为FAK抑制剂,活性最好的化合物对FAK酶(FRET)的抑制活性IC50值为0.23 μM.在实验操作中他们以诺华制药公司设计的1、2、4-三嗪类抑制剂TAE-226为对照品,发现这些化合物对癌细胞株 (U87-MG和HCT-116) 表现出较弱的细胞毒性作用,但是具有较强的抗肿瘤作用.总结所有数据的结果后,该团队强调了芳香环插入催化口袋中所产生的氮的数量和位置以及氢键和疏水相互作用的重要性.此外,活性位点内极化水分子的势效应可能在配体与大分子相互作用中会起重要作用[46].
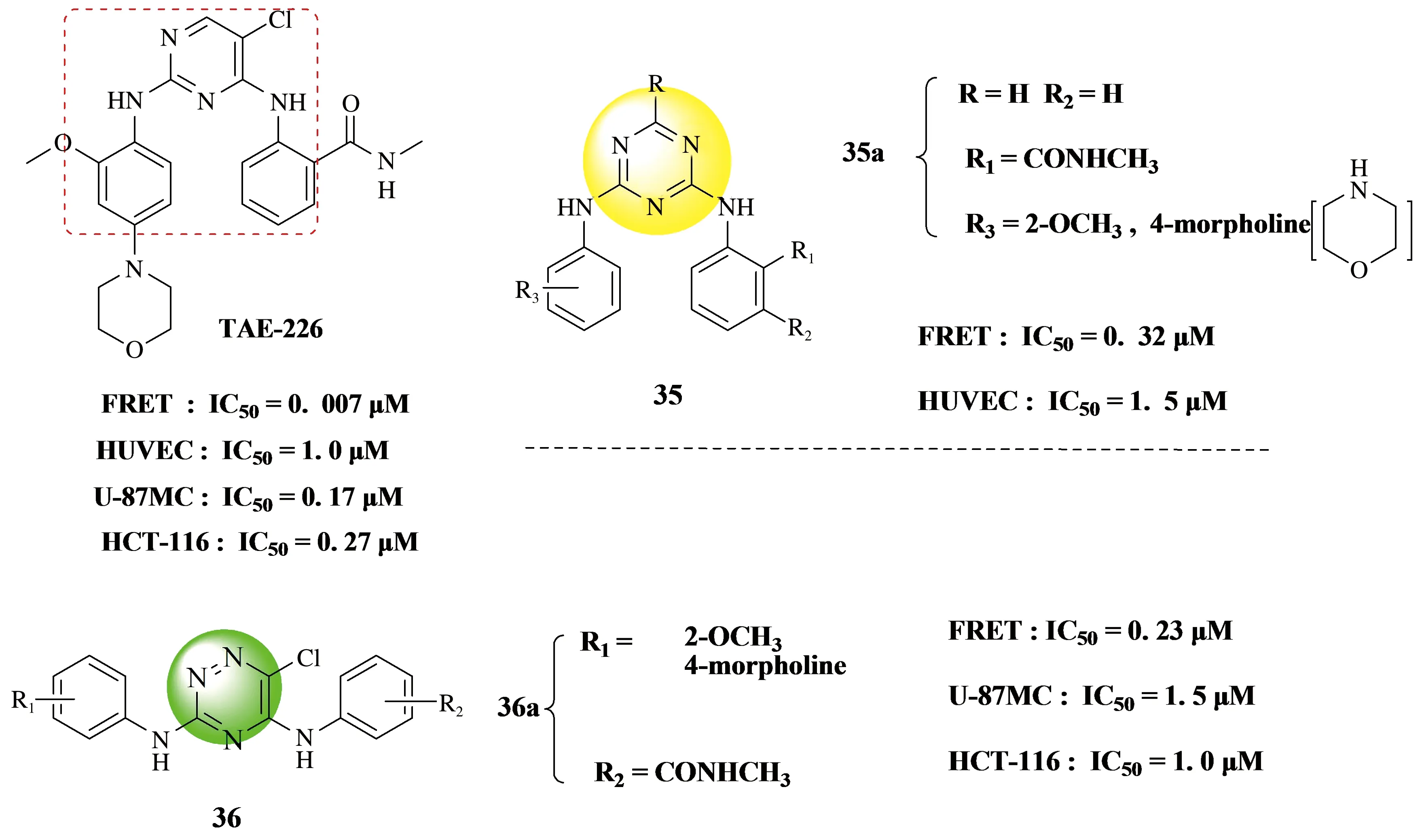
Figure 12 Pascal Dao团队合成的三嗪类FRET酶抑制剂
6 其它作用靶点
6.1 以原肌凝蛋白受体激酶(Trk)为靶点
原肌球蛋白受体激酶(Trk)是一种通过染色体重排激活的癌基因.Trk共存在三种亚型:TrkA、TrkB、TrkC,均能与具有不同特异性和亲和性的神经营养因子结合.激活的Trk受体信号通过RAS、AKT或PLC-g诱导神经突生长、细胞生长存活.因此,Trk家族已经成为治疗癌症和疼痛的重要药物靶点.此外,TrkA的药理调节也为治疗癌症以及癌症相关的疼痛和化疗耐药提供了一种新的方法[47].2014年期间,Brendan Frett团队[48]就设计合成了一系列吡嗪类TrkA抑制剂37、38、39 (见Figure 13),3类化合物中39a活性最突出.2017年期间,Vadim Bernard-Gauthier团队[49]也合成出了一系列氟喹唑啉类Trk抑制剂40 (见Figure 13),其中化合物40a抑制原肌球蛋白受体激酶(Trk)的活性最好.
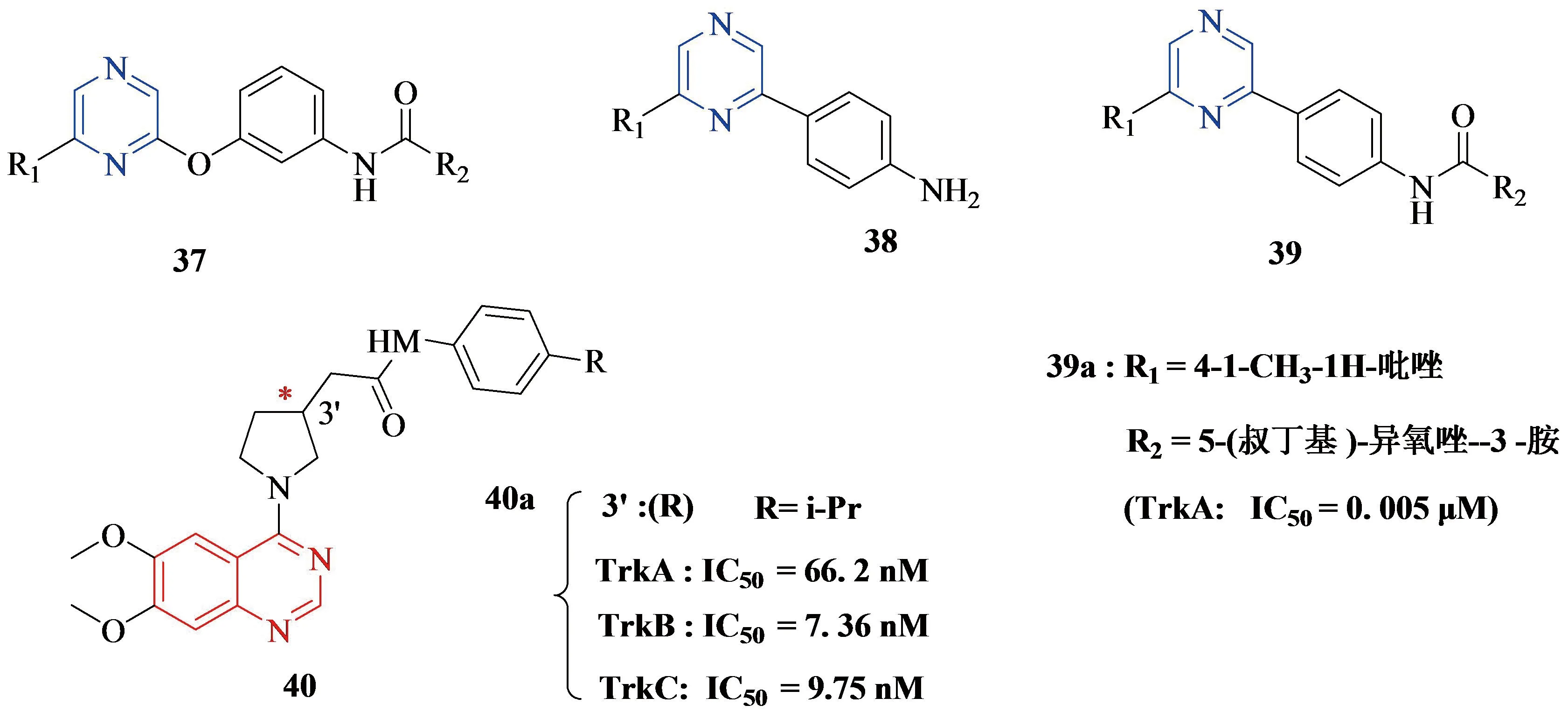
Figure 13 Frett团队和Vadim团队合成的吡嗪类和喹唑啉类Trk抑制剂
6.2 以受体酪氨酸激酶(RTK)为靶点
受体酪氨酸激酶( RTKs )已成为癌症发展的各个方面的关键调控因子,包括增殖、侵袭、血管生成和转移,RTK家族是抗癌药物开发的一个重要治疗靶点.成纤维细胞生长因子受体( FGFR )就是受体酪氨酸激酶( RTK )超家族的一个亚家族,参与调节细胞增殖和迁移、血管生成、器官发育等过程.例如:Gaozhi Chen团队[50]就于2014年合成了两个系列新型吲哚类RTK抑制剂41和42(见Figure 14),它们之间的主要区别在于连接剂之间的羟吲哚和疏水基团的差别.其中化合物42a和42b(见Figure 14)对许多癌细胞系表现出很强的活性,并对受体酪氨酸激酶c-Kit表现出明显的抑制作用,此外,细胞周期分析也证明了激酶抑制活性。
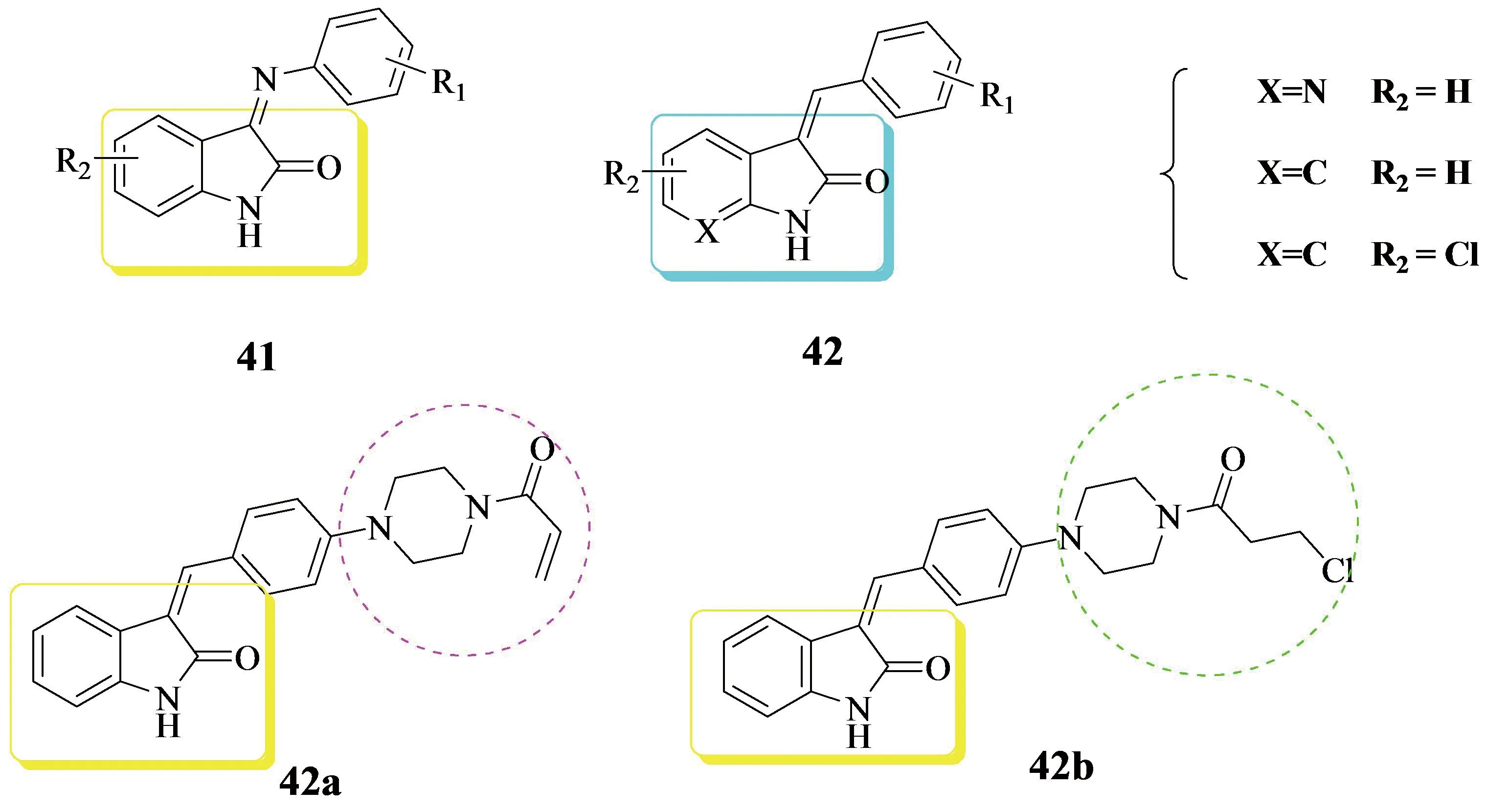
Figure 14 Gaozhi Chen团队合成的吲哚类RTK抑制剂
7 结论
本文通过查阅靶向抗血管生成的抗肿瘤药物近年来的研究进展,对具有较好抗血管生成效果的抗肿瘤活性小分子进行了综述.基于靶点和骨架结构分类的方式对其发展状况、特性和前景进行了分析和总结,旨在为后续新的抗血管生成活性分子的发现及结构优化工作提供思路.文中主要整理并列举了VEGFR-2(血管内皮生长因子受体-2),FGFR(成纤维细胞生长因子受体)、EGFR(表皮生长因子受体)、AKt(蛋白激酶)、FAK(局灶性粘附激酶) 、Trk(原肌凝蛋白相关激酶)、RTK(受体酪氨酸激酶)等7个主要靶点,并且以结构分类为依据,进一步对许多研究团队合成的小分子分别进行了阐述.综述表明了噻唑环、嘧啶环、噻吩环、吡嗪环等都是具有特征活性的杂环,并且是在开发抑制剂方面利用比较多的重要骨架.
猜你喜欢
电子乐园·上旬刊(2022年5期)2022-04-09
中国新技术新产品(2020年5期)2020-05-06
科学24小时(2018年1期)2018-01-10
农业工程技术·温室园艺(2017年3期)2017-07-13
中学生数理化·高二版(2016年3期)2016-12-26
现代养生·下半月(2016年6期)2016-10-21
汽车零部件(2016年6期)2016-07-18
安徽医科大学学报(2015年9期)2015-12-16
数理化学习·高三版(2009年3期)2009-04-30
中学生数理化·高二版(2008年2期)2008-10-19
