
低取代度磺丁基醚-β-环糊精合成及结构表征
2020-11-24 04:23赵文朋张阳倩张毅民
高校化学工程学报 2020年5期
赵文朋,张阳倩,张毅民
(天津大学 化工学院 绿色合成与转化教育部重点实验室,天津300350)
1 前 言
Villiers[1]使用环糊精-糖基转移酶(cyclodextrin-glycosyltransferase,CGTase)作用于淀粉发生酶促反应,并从得到的降解产物中分离出环糊精(cyclodextrin,CDs),其中β-环糊精(β-cyclodextrin,β-CD)是最为重要的一种[2]。β-CD是含有7个椅型构象的D-吡喃葡萄糖单元并通过α-(1,4)糖苷键首尾相连形成的一个空腔截椎体。由于其外亲水内疏水的特殊环状结构,可与许多有机和生物分子形成包合物及分子组装体系,在催化、分离、食品以及药物等领域中得到广泛应用[3-6]。然而,β-CD 母体溶解度低并具有肾毒性,在应用上受到限制[7-8]。为了突破这些限制,对β-CD的基团进行改性形成衍生物,相对于β-CD具有较小的毒性从而可以扩大β-CD应用范围[9-13]。
现有的衍生物中,负电性的磺丁基醚-β-环糊精(sulfobutyl ether-β-cyclodextrin,SBE-β-CD),如图1所示。因其卓越的水溶性和生物相容性被广泛应用于药物制剂[14-16]、分子识别[17]等领域。SBE-β-CD 的合成工艺常采用1,4-二氧六环或四氢呋喃作反应介质,溶剂价格昂贵并具有一定毒性,而且产物的收率也较低[8,18]。Song 等[19]在碱溶液中采用“一锅法”合成了磺烷基醚-β-环糊精,Ma 等[20]和张毅民等[21]采用“三步法”合成SBE-β-CD,避免了使用有毒溶剂,但仍存在工艺过程中加碱及反应后脱盐等繁杂工序,不仅给生产造成不便,而且会带来盐、碱环境污染和资源浪费等问题[19-21]。碱性电解水是一种带有多种功能的离子水,具有碱性特征且在使用后可以自然恢复到水的状态[22],但它在合成工艺中的应用却鲜有提及。本文试图以不同pH的碱性电解水为介质,通过β-CD和1,4-丁磺内酯在微波辐射反应器中反应合成SBE-β-CD,发展β-CD在新型介质中的反应体系。
2 实验(材料与方法)

图1 SBE-β-CD结构图Fig.1 Structure of SBE-β-CD
2.1 材料与仪器
Bruke液体核磁共振仪(400 MHz);Thermo Fisher 液相色谱质谱联用仪;Nicolet 傅里叶变换红外光谱分析仪;Vario Micro Cube元素分析仪;微波反应器(上海丞武仪器科技有限公司);FE28 pH 计(梅特勒-托利多仪器有限公司)。
1,4-丁磺内酯(分析纯,郑州阿尔法化工有限公司);β-CD(分析纯,天津市科密欧化学试剂有限公司);碱性电解水(pH=13.1,天津日望环境技术有限公司);D2O(萨恩化学技术有限公司);其余均为市售分析纯试剂。
2.2 实验方法
将10 g 重结晶后的β-CD加入盛有158~280 mL 不同pH 的碱性电解水的三口烧瓶中,然后放入带有三口烧瓶上端连接滴液漏斗和冷凝管的微波反应器内,将微波反应器功率设置为600 W,温度设定为75℃,将β-CD加热至全部溶解并达到设定温度后在该功率下维持0.5 h,然后通过滴液漏斗缓慢滴加2.05~3.27 mL 的1,4-丁磺内酯,滴加完毕后,反应在75℃时持续2~3 h。反应结束后,待反应混合物冷却至室温,将其暴露在空气中不断搅拌,使反应混合物pH呈中性并用LNG-NF-101纳滤膜分离其中的小分子杂质,将所得溶液干燥,得到白色粉末状SBE-β-CD,将其称重计算收率。
3 结果与讨论
3.1 反应机理
电解水是在装有稀释盐水的电解池中电解得到,在电解池两端的电解槽中阳极和阴极分别得到酸性电解水和碱性电解水,阳极和阴极的反应式如(1)、(2)、(3)所示[23]:

阴极电解出的碱性电解水pH 在13左右,具有明显碱性特征。SBE-β-CD的合成是典型的不对称醚键的醚化反应,符合SN2亲核取代反应机理。β-CD包含3种类型的羟基,分别为伯羟基(C6─OH)和仲羟基(C2、C3─OH),3种类型的羟基具有不同的亲核性,反应活性顺序为C6─OH >C2─OH > C3─OH。以不同pH 的碱性电解水作为反应介质,β-CD中C2位和C6位上的羟基都会发生取代反应,1H NMR 谱图中C1位上的部分氢原子化学位移向低场移动说明C2位上的羟基发生了取代反应,13C NMR 谱图中C6、C5位上的化学位移部分向低场移动,其他位上碳的化学位移变化很小,这说明C6位上的羟基也发生了取代反应。氢氧根离子诱导β-CD(1a)生成烷氧基负离子化的β-CD(2a),由于β-CD和氢氧根离子的反应是可逆的动态过程,因此烷氧基负离子(4a)不断生成并作为亲核试剂去攻击1,4-丁磺内酯(5a)环上与氧相连的活性C原子,C─O键断裂的同时烷氧负离子与C原子结合形成新的C─O键(6a),最终生成SBE-β-CD(3a)。反应流程和反应机理如图2、3所示[24]。
3.2 反应条件对反应的影响
3.2.1原料配比对反应的影响
采用不同pH 的碱性电解水代替氢氧化钠溶液作反应介质和催化剂[24-25],通过改变β-CD和1,4-丁磺内酯摩尔配比,利用微波辐射的热效应和分子场效应,实现反应过程。不同取代度产物的反应物用量及收率如表1所示。由表1可见,当1,4-丁磺内酯摩尔配比是β-CD的2~10 倍时,都可以得到相应SBE-β-CD产物,当两者摩尔配比为2~3倍时,分别得到2-取代和2.5-取代产物,当两者摩尔配比为4时,主要得到3-取代产物,在这一阶段产物的收率基本保持在70%~75%,然而,随着1,4-丁磺内酯摩尔配比增加,5-取代、6-取代和7-取代的产物逐渐减少,因此,不适合合成平均取代度为5以上的产物。
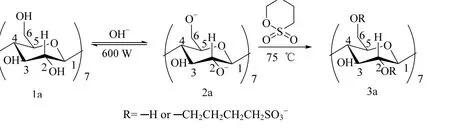
图2 SBE-β-CD的反应流程Fig.2 Reaction path of SBE-β-CD
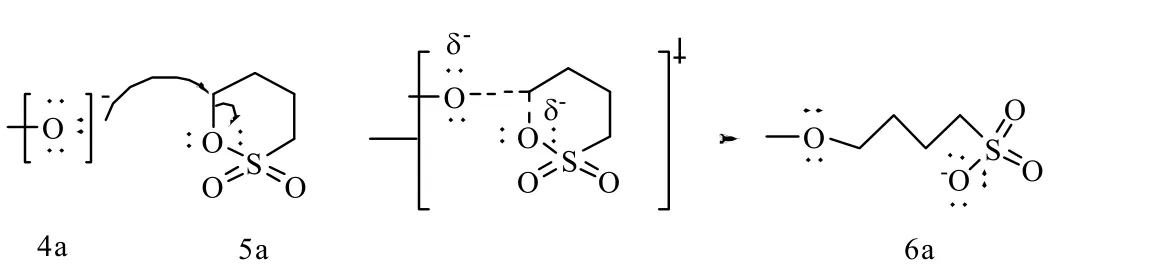
图3 SBE-β-CD的反应机理Fig.3 Reaction mechanism of SBE-β-CD

表1 不同取代度产物的反应物用量变化及收率Table 1 Yield of SBE3-β-CD under different reactant ratiosand substitution degrees
3.2.2传统水浴法与微波辐射法对反应的影响
以合成SBE3-β-CD为例考察了传统水浴法与微波辐射法对反应的影响。当β-CD为10 g 和1,4-丁磺内酯为3.27 mL(1,4-丁磺内酯与β-CD摩尔配比为4)时,在碱性电解水pH=12.7,分别用传统水浴法和微波辐射法在75℃下进行反应,同时与在氢氧化钠水溶液中反应进行对比,水浴法与微波辐射法对SBE3-β-CD收率的影响如表2所示。由表2可见,在碱性电解水中,无论是采用传统水浴法,还是微波辐射法,所得产物的收率都比在氢氧化钠水溶液中要低;采用微波辐射法,无论是利用碱性电解水,还是氢氧化钠水溶液,反应时间都大大缩短。这可能是由于微波的场效应诱导和强化反应,活化了分子,降低了反应自由能,使反应由16~19.5 h 缩减为2.5 h,大大提高了反应速率。

表2 水浴法与微波辐射法对SBE3-β-CD收率的影响Table 2 Yield of SBE3-β-CDby water bath heating and microwave heating
3.2.3不同pH的碱性电解水对SBE-β-CD收率的影响
以合成SBE3-β-CD为例考察不同pH 的碱性电解水对产物收率的影响。取定量的pH=13.1的碱性电解水,将其稀释至不同的pH 值作为碱性介质,75℃反应2.5 h,产物收率如表3所示。由表3可见,当碱性电解水的pH为12.7时,SBE-β-CD的收率最高。这可能是因为pH 低于12.7时β-CD羟基上的O─H 键断裂较少,烷氧基负离子化的程度小,烷氧基负离子浓度限制了产物的生成,收率较低,pH 高于12.7会将加入的1,4-丁磺内酯分解为4-羟基丁磺酸盐等副产物,原料的减少影响了产物收率的提高。制备其他低取代度SBE-β-CD应以此碱性电解水pH为依据进行。
3.3 产物的表征
3.3.11H NMR 表征
为了分析产物的结构特征,以重水为溶剂分别对β-CD和SBEn-β-CD进行核磁氢谱表征,其表征结果如图4所示。由图4可见,在β-CD的图谱中,化学位移δ=5.01处的峰归属于C-1位上的7个氢,δ=4.71处的峰归属于溶剂重水上的氢,δ=3.47~3.93处的峰归属于β-CD骨架上的氢。对比SBEn-β-CD和β-CD的1H NMR 谱图,在SBE-β-CD中可以观察到在δ为2.74和1.87的位置各出现了一个新峰,经分析,两个新峰分别归属于取代基团─CH2─CH2─CH2─CH2─SO3-上靠近SO3-的─CH2─上的2个氢和取代基团中心─CH2─CH2─上的4个氢,且在δ为2.74与1.87处峰的积分比为1:2,符合理论计算值,说明磺丁基基团已经取代到β-CD上。取代基团中靠近醚键的─CH2─上的两个氢与β-CD骨架上的氢重叠于δ=3.47~3.93处,不易辨别。1H NMR 还可以提供关于取代基的取代位置的信息,在SBEn-β-CD谱图中C-1处的氢峰为两个,分别在δ为5.10和5.01处,比β-CD上C-1处氢的化学位移数值要大,向低场移动,这是因为C-2位上的羟基被取代后,取代基团─CH2CH2CH2CH2SO3-的去屏蔽效应作用于C-1位上的氢使其部分向低场移动[26],这表明磺丁基基团取代到β-CD上。

表3 碱性电解水 pH 对 SBE3-β-CD 收率的影响Table 3 Yield of SBE3-β-CD under different pH of alkalineelectrolyzed water

图5 SBE3-β-CD核磁共振碳谱图Fig.5 13C NMR spectra of SBE3-β-CD

图4 β-CD 和SBE n-β-CD的核磁氢谱图Fig.4 1H NMR spectra of β-CD and SBE n-β-CD(a) β-CD (b) SBE2-β-CD (c) SBE2.5-β-CD (d) SBE3-β-CD
3.3.213C-NMR 表征
以重水为溶剂对SBE3-β-CD 进行13C-NMR 表征,表征结果如图5 所示。碳谱数据为δ(×10-6):101.4(C-1),80.5(C-4),72.2(C-5),72.1、71.9(C-3,2),60.8(C-6),60.2(C-1’),50.1(C-4’),28.9(C-2’),21.0(C-3’)。由数据可以看出C-1,C-4,C-6的化学位移比较分散,C-2,C-3,C-5的信号由于重叠导致其化学位移比较集中,这些峰的位移值和β-CD的相同,说明SBE-β-CD拥有和β-CD同样的框架结构。SBE-β-CD在δ=28.9,21.0,50.1处产生3个新的特征峰,分别归属于取代基─CH2─CH2─CH2─CH2─SO3─上的C-2’,C-3’,C-4’,表明磺丁基基团已经接枝到β-CD上,成功合成了SBE-β-CD。此外,C-6位上的化学位移向低场移动了9.2×10-6,邻位C-5位上的化学位移向低场移动了4.8×10-6,其他位碳上的化学位移值变化很小,表明在C-6位上的羟基发生了取代反应。
3.3.3 FT-IR 表征
红外光谱可以用来鉴别β-CD和SBEn-β-CD的结构,从而推测产物的化学结构信息。β-CD及各取代度产物红外光谱如图6所示。由图6可见,几种衍生物和β-CD具有类似的红外图谱,且β-CD和SBEn-β-CD的特征红外光谱数据为3 420、2 935、2 890、1 644、1 173、1 037、605、537 cm-1。537 cm-1处的吸收峰为S─O伸缩振动峰,且该峰强度随取代度升高而增大。605 cm-1处出现尖锐吸收峰,该峰归属于C─S伸缩振动峰。1 644 cm-1为环糊精空腔中少量水形成的分子内键合水特征峰,2 890和2 935 cm-1分别是─CH2─的对称和不对称伸缩振动特征峰。3 420 cm-1处为O─H伸缩振动形成宽缔合峰,在SBE-β-CD中,随着取代度的增大,羟基数量减少,该峰峰宽也逐渐变窄,说明羟基发生了取代反应。1 173、1 037 cm-1是吡喃糖环上C─O的伸缩振动峰,此外,SBE-β-CD中磺丁基与烷氧基新形成的C─O─C在1 173 cm-1的伸缩振动峰与吡喃糖环上C─O伸缩振动峰重合,与β-CD相比,SBE-β-CD在此处的峰形变宽,这些都表明合成了SBE-β-CD。
3.4 产物平均取代度计算
3.4.1核磁氢谱法
1H NMR 谱图除了分析产物的结构,它还可以用来计算产物的平均取代度。在SBE-β-CD中δ为2.74和1.87左右的位置各有一个峰,两个峰分别归属于取代基团─CH2CH2CH2CH2SO3-上靠近─SO3-的─CH2─上的2个氢和取代基团中心─CH2─CH2─上的4个氢。氢谱法计算平均取代度可用δ=5.20~4.95(C-1上的7个氢)处的峰面积之和与δ=2.74(取代基团中靠近─SO3-的─CH2─上的2个氢)处的峰面积的比值来表示。如式(4)所示:

图6 β-CD和SBE n-β-CD的红外光谱图Fig.6 Infrared spectra of β-CD and SBE n-β-CD(a) β-CD(b)SBE2-β-CD(c)SBE2.5-β-CD(d)SBE3-β-CD

式中:DS为产物平均取代度,A1为δ=5.20~4.95处积分峰面积,A2为δ=2.74处积分峰面积[27]。
对SBE3-β-CD进行核磁氢谱表征,表征结果如图7所示。由图7可见,产物A1峰面积为1.18,A2峰面积为1.00,经计算可得DS=2.97。
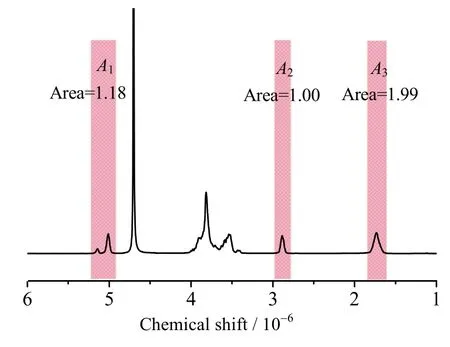
图7 SBE3-β-CD的积分图Fig.7 1H NMR analysis of SBE3-β-CD

表4 质谱峰理论结果数据Table 4 Theoretical results of massspectrometry peaks
3.4.2质谱法
采用负离子源的电喷雾质谱仪对SBE-β-CD进行表征,碎片离子为带多个负电荷的负离子[28]。质谱峰理论结果数据如表4所示。质谱法计算产物的平均取代度,主要是利用质谱中各个碎片离子峰所代表的取代度及相应的丰度进行计算,计算方法如式(5)所示[25,29]:

式中:n为碎片离子峰所代表的取代度,An为碎片离子峰的丰度。
对SBE3-β-CD进行质谱表征,其表征结果如图8所示。由图8可见,MS(ESI)m/z(%):1 269.4(3.78,n=1)、781.7(8.83,n=3)、770.2(2.68,n=3)、702.2(66.0,n=2)、565.8(7.08,n=4)、558.5(1.96,n=4)、513.1(100,n=3)、458.1(1.93,n=5)、361.9(9.50,n=5),由此可知SBE3-β-CD是取代度1~5 的混合物,各个取代度的相对丰度分别为SBE1(3.78%),SBE2(66.00%),SBE3(111.51%),SBE4(59.07%),SBE5(11.43%),由其取代度和丰度计算可得产物平均取代度DS=3.03,与氢谱计算的DS基本一致。
3.4.3元素分析法
利用元素分析仪检测出SBE3-β-CD中元素(C、H、S)的质量分数w见表5,根据C、S元素的质量分数来测定产物的平均取代度,计算方法如式(6)所示:
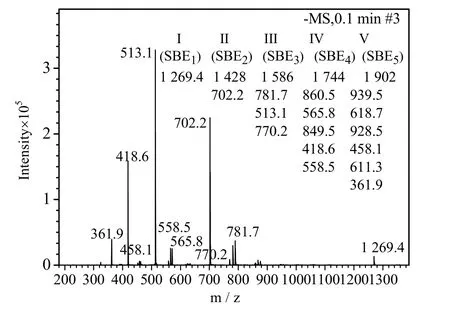
图8 SBE3-β-CD的质谱图Fig.8 Mass spectrum of SBE3-β-CD

表5 SBE3-β-CD的元素分析结果Table 5 Elemental analysis of SBE3-β-CD

式中:MC和w(C)分别表示C的相对原子质量和产物中C的质量分数,MS和w(S)分别表示S的相对原子质量和产物中S的质量分数[8]。
经计算,产物的平均取代度DS=2.62。
3.4.4各方法计算取代度对比
分别利用核磁氢谱法、质谱法、元素分析法测定产物的平均取代度并对不同方法测定的结果进行了对比。3种方法测定的取代度结果有所差异,核磁氢谱法和质谱法测定的取代度分别为DS=2.97和DS=3.03,元素分析法测定的取代度为DS=2.62,低于前两种方法的结果。这可能是由于产物取代度较低,核磁氢谱法和质谱法因为低取代度产物的溶解度限制,使得计算出的DS偏高,元素分析法测定由于不需样品溶解从而避免这一误差,所以元素分析法计算出的取代度DS可信度最高。
4 结 论
(1)控制β-CD 和1,4-丁磺内酯摩尔比为1:(2~4)和不同pH 的碱性电解水加入量,在75℃反应2.5 h,合成了平均取代度分别为2、2.5、3的SBE-β-CD;且当反应介质的pH=12.7时,产物收率达到70.1%。微波辐射法较水浴法,反应时间可缩短13.5~17 h,收率由62.4%提高到70.1%。
(2)以不同pH的碱性电解水为介质,利用微波辐射协同作用合成低取代度SBE-β-CD工艺具有反应条件温和、后处理过程简单的特点,是一种环境友好的技术工艺路线。
猜你喜欢
长江蔬菜(2022年18期)2022-10-27
长江蔬菜(2022年4期)2022-02-16
现代农村科技(2020年6期)2020-12-20
上海建材(2020年12期)2020-04-13
中成药(2018年8期)2018-08-29
中成药(2018年6期)2018-07-11
铜仁学院学报(2018年6期)2018-07-05
中成药(2018年4期)2018-04-26
中成药(2017年5期)2017-06-13
浙江理工大学学报(自然科学版)(2015年5期)2015-03-01
