
RASGRP2基因罕见突变导致遗传性血小板功能障碍1例报道及文献复习*
2020-11-21 03:27:36庹媛媛
华中科技大学学报(医学版) 2020年5期
庹媛媛, 金 皎
贵州医科大学附属医院儿科血液专科,贵阳 550001
遗传性血小板功能障碍(inherited platelet function disorders,IPFD)是一种多伴常染色体遗传的罕见出血性疾病,儿童多见,典型表现为不同程度的出血,以皮肤黏膜出血、月经过多、外伤后难以止血多见,伴或不伴血小板减少[1]。随着分子生物学技术的发展,二代测序在临床的应用,发现了越来越多与血小板功能有关的基因突变。其中RASGRP2基因编码钙和甘油二酯调节鸟嘌呤交换因子Ⅰ(CalDAG-GEFⅠ),参与血小板活化过程中的信号转导,影响血小板粘附、聚集功能,其功能缺陷可导致出血性疾病的发生[2]。目前国内外有少量相关基础研究及病例报道。本文报道1例RASGRP2基因罕见双重杂合突变并可能因此导致的出血性疾病病例,进行相关文献复习,以期提高对遗传性血小板功能障碍临床诊断及治疗的认识。
1 临床资料
1.1 病史及体格检查
患儿,男,4岁,因“反复鼻出血3年余”于2018年1月来我院儿科血液专科门诊就诊。患儿1岁时出现自发性鼻腔出血,不易止血,皮肤、关节等其他部位无显著出血,随后反复鼻腔出血,每周1~2次到每月2~3次不等。病程中合并小细胞低色素性贫血。外院多次诊断鼻出血,予鼻腔填塞止血及口服铁剂治疗。查体:轻度贫血貌,全身皮肤无瘀点、瘀斑,全身浅表淋巴结未触及肿大,睑结膜苍白,双侧鼻腔可见血痂附着,心肺腹查体无特殊,四肢无畸形。既往史、个人史阴性,否认父母近亲结婚史及出血性疾病家族史。
1.2 外院辅助检查回顾
血常规:血红蛋白波动于60~112 g/L,呈小细胞低色素,血小板计数、血小板平均体积均正常。血块收缩试验、纤维蛋白原、凝血因子Ⅱ、Ⅴ、Ⅶ、Ⅷ、Ⅸ、Ⅹ、Ⅺ、Ⅻ活性筛查及vWF检测均正常。地中海贫血基因检测阴性。血小板功能检测:血小板对ADP聚集率18.5%。骨髓常规:增生明显活跃,粒细胞∶红细胞=2.97∶1,红系增生,成熟红细胞大小不均,以小细胞为主,中心淡染区扩大,全片可见468个巨核细胞,血小板数量轻度增高,散在分布,细胞内铁0%,细胞外铁阴性,提示缺铁性贫血骨髓像。采取外周血标本送检武汉康圣达医学检验所,应用二代测序筛查遗传性血液病相关近700个基因,结果显示患儿第11号染色体上RASGRP2基因双重杂合突变:64497534位点[c.1545C>A,p.515C>X](RASGRP2 p.515C>X),64509585位点[c.74-1G>C]。
1.3 家系验证
利用一代Sanger测序验证先证者及父母、弟弟RASGRP2上述突变位点的情况(图1)。患儿母亲检出RASGRP2基因64497534位点与先证者相同的点突变,为该突变杂合携带者,64509585位点未检测出突变;患儿父亲,检出RASGRP2基因64509585位点与先证者相同点突变;患儿弟弟,7月龄,未检出RASGRP2基因突变,为野生型。上述家庭成员均无过度出血表现,家系图谱见图2。应用SIFT、PolyPhen2、LRT、Mutation Taster方法进行突变分析,在NCBI上比对NM_001098670、NM_001098671、NM_153819等3种转录本,64497534位点突变位于13号外显子,为无义突变[c.1545C>A,p.515C>X],64509585位点突变位于4号外显子,为剪接突变[c.74-1G>C],通过查询千人基因组计划数据库、人类遗传突变数据库、美国国家生物技术信息中心单核苷酸多态性数据库等国际多个数据库,暂未发现上述位点突变的人类相关致病报道。
1.4 诊断及治疗、随访
结合患儿临床表现、血小板功能检测,以及二代测序RASGRP2基因双重杂合突变结果。该患儿诊断为遗传性血小板功能障碍(IPFD)及缺铁性贫血。医嘱:①注意外伤,避免鼻腔出血;②铁剂治疗,每月监测血常规+网织红细胞;③儿科血液专科门诊随诊。随访:患儿出院后遵医嘱,日常生活注意避免出血发生。5岁后鼻出血发生频率下降,约1~2次/月,贫血纠正。治疗过程中未予输注血小板等特殊治疗。
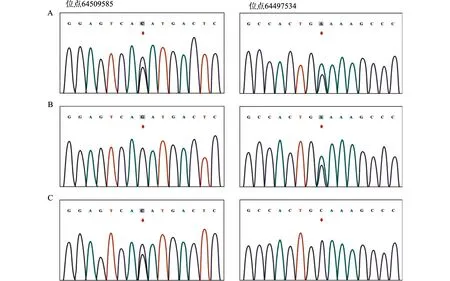
A:先证者64509585位点[c.74-1G>C],64497534位点[c.1545C>A];B:母亲64509585位点未检测出突变,64497534位点[c.1545C>A];C:父亲64509585位点[c.74-1G>C]突变图1 先证者及父母RASGRP2基因突变位点Sanger测序图Fig.1 Sanger sequencing analysis of the patient and his parents with the mutations in RASGRP2
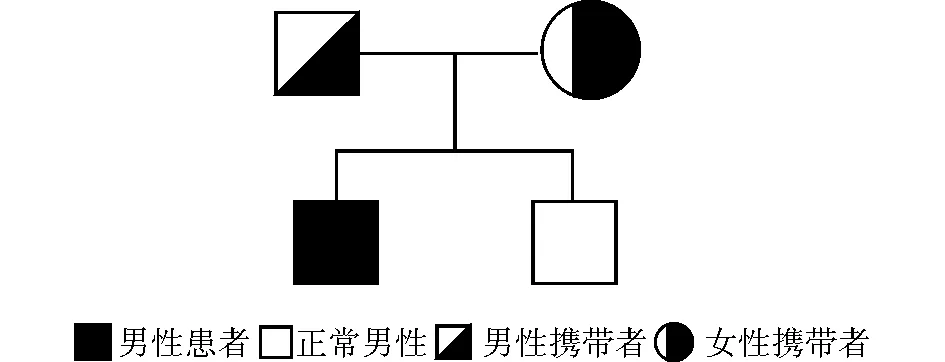
图2 先证者家系图谱Fig.2 Family pedigrees of the patient
2 讨论
人类的出凝血系统,由血管、血小板、凝血系统、抗凝纤溶系统共同调节,其中任何一个因素的异常,将导致患者发生过度出血或血栓形成。在儿童患者中,血小板数量的减少、先天性或获得性凝血因子缺乏及血管因素,是最常见的出血性疾病的病因,血小板功能障碍性疾病相对少见。临床上将血小板功能障碍性疾病分为遗传性和继发性,继发性血小板功能障碍性疾病多由于其他血液病和非血液病使用某些药物影响血小板功能所致。小儿以遗传性血小板功能障碍性疾病多见,遗传性血小板功能障碍是一种多数伴常染色体遗传的罕见疾病,全球散发,是一组和不同严重程度出血有关的异质性疾病,多数是由于参与血小板粘附、聚集、分泌和信号转导等功能的基因缺陷导致的一类血小板疾病。以血小板无力症(Glanzmann thrombasthenia,GT)及巨大血小板综合征(Bernard-Soulier syndrome,BSS)最常见。临床以皮肤黏膜自发出血、外科手术和创伤后出血不止为主要表现。2015年国际血小板生理学科学小组委员会发布IPFD诊断指南[3],指出该病诊断主要依赖于病史和查体、实验室对血小板计数和形态的评估、血小板功能的评估以及流式细胞仪检测血小板颗粒和表面标记物含量,疑似病例进一步完善二代测序(NGS)。随着二代测序的应用,越来越多的IPFD借助二代测序获得诊断,该病已不再像之前所认为的那样罕见。现已被认可有51种基因与该病的临床表现及实验室检查结果相关[4]。
RASGRP2基因位于11号染色体长臂13区1带,1998年首次被克隆,曾被命名为人类CDC25类似物(HCDC25L)、Ca2+及二酰甘油调节的鸟嘌呤交换因子Ⅰ(CalDAG-GEFⅠ)。其编码表达的CalDAG-GEFⅠ蛋白在造血系统的血小板、中性粒细胞、T淋巴细胞中强烈表达[5-6],是一种膜结合的信号转导蛋白,可整合钙离子和二酰基甘油(DAG)通路,其位于胞质内的短片段与Rap1和Rap2结合,通过活化Rap,调控细胞对外界刺激的反应。Bergmeier等[7]研究发现敲除RASGRP2基因的小鼠其血小板Rap1活化功能存在缺陷,导致血小板信号传导功能下降,对诱聚剂反应下降,整合素介导的血小板粘附减少,血栓形成增多。Hisashi等[8]研究显示RASGRP2的缺陷可阻断蛋白激酶C(PKC)的活化,阻止整合素αⅡbβ3活化,从而影响血小板聚集、粘附的功能,导致出血性疾病的发生。由此可见,CalDAG-GEFⅠ对血小板活化的关键事件——PKC活化释放和整合素αⅡbβ3的激活调节均十分重要,其缺陷影响正常血小板的粘附、聚集功能[9]。由RASGRP2缺陷所导致的出血性疾病和某些血小板无力症(GT)发病机制相似,实验室显示血小板LTA对常见诱聚剂反应下降,但血小板表面aⅡbβ3表达仅轻度或中度下降[10-11]。Canault等[12]报道RASGRP2第8外显子的纯合c.742G-T突变,引起CDC25催化结构域的结构保守区域发生了G248W取代,导致RasGRP2功能缺陷,血小板内外信号传导缺陷,干扰血小板聚集和扩散。这是关于RASGRP2突变体导致遗传性血小板功能障碍的首次报道。目前人类遗传突变数据库(HGMD)收录RASGRP2基因24个突变中,有20个是点突变,3个是缺失突变,1个是重复突变。近期,Westbury等[13]研究者通过对2042例不明原因的出血患者家系进行高通量测序和表型数据分析,又发现11例新的RASGRP2双等位基因变异可能具有致病性。目前临床表型-基因型分离验证存在难度,将基因突变与潜在的出血性疾病联系起来仍是一个持续性的挑战。
本报道通过对CalDAG-GEFⅠ蛋白质结构的分析发现,先证者RASGRP2基因64497534位点突变[c.1545C>A,p.515C>X],位于13号外显子二酰甘油结合结构域(C1),无义突变导致终止密码子提前编码,阻断了肽链的合成;64509585位点[c.74-1G>C]突变位于4号外显子前一个碱基,4号外显子位于Ras交换基序(REM)。通过human splicing finder软件预测c.74-1G>C剪接受体位点结构,显示先证者剪接位点被破坏,导致mRNA剪接异常,无法正常表达RAS交换基序,从而影响下游蛋白的表达。由此可以预测先证者的双重杂合突变影响CalDAG-GEFⅠ结构,是可能致病的(图3)。
IPFD的临床出血表现存在异质性,及时的诊断及分类,评估病情预测出血程度对该类患者尤为重要。输注血小板被认定为目前治疗严重出血或手术预防出血的常用措施,产生抗血小板抗体是常见的不良反应,为减少同种免疫反应,这类患者应该输注去白细胞或者HLA匹配的血小板[14];许多临床医师鉴于此不良反应,更倾向选择重组凝血因子Ⅶa(rFⅦa)。Di Minno等[15]研究者指出单纯用重组凝血因子Ⅶ治疗血小板无力症止血,成功率可达91.0%;全球已有散在的造血干细胞移植治疗IPFD成功的病例,但其可能出现严重并发症等情况,被慎重考虑。此外,去氨加压素(DDAVP)、激素避孕药以及抗纤维蛋白溶解疗法也被视为有效的辅助治疗方法。目前遗传性血小板功能障碍无有效的靶向治疗措施,基因治疗总体处于动物实验阶段,且高额的费用,短时间可能难以在临床实施。值得注意的是,所有患者均有可能合并缺铁性贫血,在治疗中需得到关注。预防仍是原发性出血的最佳管理方法,因此需注意口腔卫生,避免高危活动,并适当早期干预。
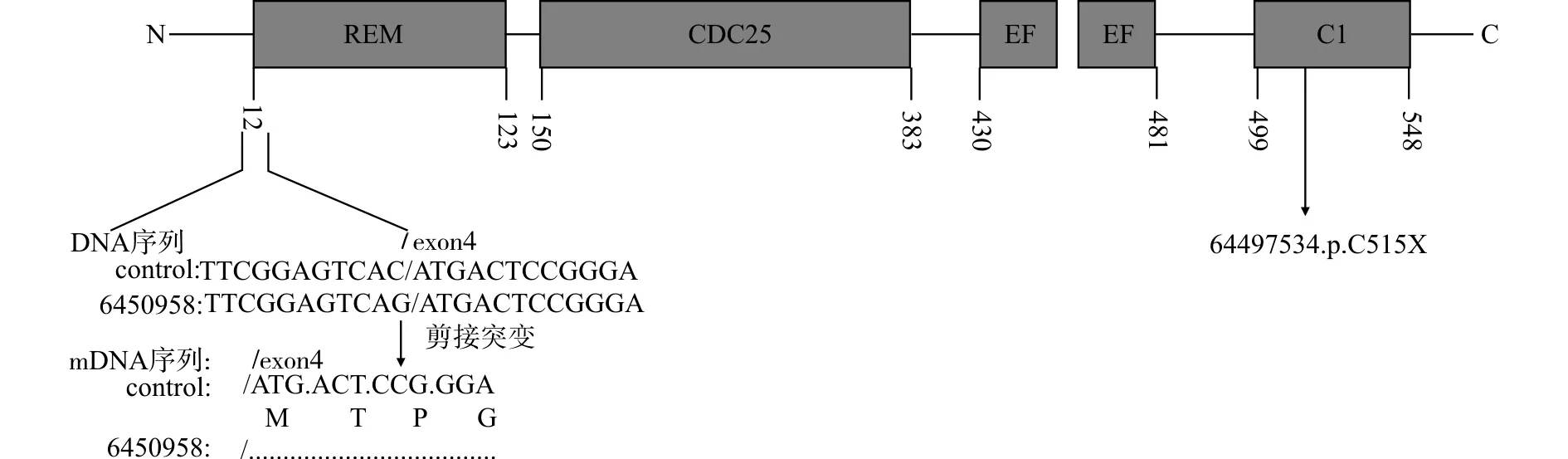
REM:RAS交换基序;CDC25:催化结构域;EF:钙结合位点;C1:二酰甘油结合结构域图3 RASGRP2基因[c.74-1G>C]与[c.1545C>A]突变在CalDAG-GEFI蛋白结构中的定位与预测图Fig.3 Localization and prediction map of the[c.74-1G>C]and[c.1545C>A]mutations within the RASGRP2 sequence and the encoded protein CalDAG-GEFI
综上所述,RASGRP2基因缺陷通过影响血小板Rap1的激活,从而影响PKC及αⅡbβ3整合素的激活,导致血小板粘附、聚集功能障碍,临床表现可出现不同程度的出血。它是遗传性血小板功能障碍的致病基因之一,目前全球仍然陆续发现与疾病相关的未知遗传变异。遗传性血小板功能障碍临床表现存在异质性,实验室诊断难以达到标准化,诊断相对困难,临床上只有少部分通过二代测序明确诊断。本文首次报道RASGRP2基因罕见双重杂合突变可能导致遗传性血小板功能障碍1例,对于IPFD的认识不足导致该患儿诊断过程欠系统。对于疑似IPFD的患者应及时完善血小板功能评估,流式细胞仪检测血小板膜表面蛋白表达以及二代测序是有必要的。
猜你喜欢
中国现代医生(2022年19期)2022-11-04 10:13:29
临床输血与检验(2022年3期)2022-06-22 02:52:50
昆明医科大学学报(2022年4期)2022-05-23 13:04:50
基层中医药(2021年8期)2021-11-02 06:24:54
郑州大学学报(医学版)(2019年3期)2019-06-03 06:19:32
传染病信息(2019年2期)2019-05-17 13:16:04
中国生殖健康(2019年12期)2019-01-07 01:54:30
分子影像学杂志(2015年3期)2015-12-04 03:29:03
西南军医(2015年3期)2015-04-23 07:28:34
重庆医学(2015年12期)2015-03-05 05:52:54
