
Alstrom 综合征1例报告及文献复习
2020-11-20 12:36魏莹刘戈力郑荣秀牛婧娅
天津医科大学学报 2020年6期
魏莹,刘戈力,郑荣秀,牛婧娅
(1.天津医科大学总医院儿科,天津300052;2.天津医科大学研究生院,天津300070)
Alstrom 综合征(Alstrom syndrome, ALMS) 是一种罕见的常染色体隐性遗传病, 由Alstrom 于1959年首次报道[1]。该病发病率约为1/100 万~9/100 万,无性别差异,近亲婚配后代发病率显著增加[2]。迄今全球报道ALMS 在950例以上[3],但国内鲜有报道。该病发病机制尚不明确,缺少有效治疗方法,预后极差。本文分析天津医科大学总医院儿科收治的1例确诊ALMS 患儿的临床资料,结合国内外文献,探讨该病主要临床特征、诊断及遗传学机制。
1 临床资料
患儿,男,11 岁2个月,因近1 周发现血糖明显升高就诊。患儿为足月顺产,第1 胎第1 产,围生期未发现异常。自幼肥胖,智力低下,5 岁时被诊断为视力残疾(一级)并眼球震颤。9 岁起颈部皮肤逐渐变黑。10 岁时发现高胰岛素血症、高脂血症。除此之外,患儿还表现有间歇性泡沫尿。患儿奶奶有糖尿病病史,否认家族中其他成员糖尿病、肾脏疾病、高血压、高脂血症等疾病史(图1)。

图1 患儿一般情况
入院体格检查:肥胖体型,身高156.8 cm,体重64 kg,腰围88.5 cm,体重指数(BMI)25.62 kg/m2;体温36.4℃,脉搏90 次/min,呼吸24 次/min,收缩压/舒张压:110/70 mmHg(1 mmHg=0.133 kPa),全身皮肤较黑,面部痤疮明显,眼睑无水肿,视力减退,眼球震颤。嗅觉及听力正常。颈部、腋下、腹股沟可见黑棘皮,双乳:左侧乳核2.5 cm×2.5 cm ,右侧乳核2.5 cm×2.5 cm。心率90 次/min,心音有力,律齐,未及杂音。腹软,腹部皮下脂肪厚,肝脾肋下未触及,肾区无叩击痛。性发育Tanner 分期Ⅲ期。
入院实验室检查:血气分析电解质正常。谷丙转氨酶234 IU/L(5~40 IU/L),谷草转氨酶48 IU/L(10~40 IU/L),谷氨酰转肽酶234 IU/L(7~49 IU/L);总胆固醇4.59 mmol/L(3.59~5. 17 mmol/L),甘油三酯3.27 mmol/L(0.57~1.71 mmol/L),高密度脂蛋白-胆固醇0.75 mmol/L(0.8~2.2 mmol/L),低密度脂蛋白-胆固醇2.35 mmol/L(1.33~3.36 mmol/L)。尿β2 微球蛋白0.7 mg/L(0.1~0.3 mg/L)、24 h 尿微量白蛋白136.9 mg/24 h(0~30 mg/24 h)、24 h 尿白蛋白定量667.8 mg/24 h(30~150 mg/24 h),均升高;血尿素、肌酐未见异常,胱抑素C 1.15 mg/L(1.15 mg/L)。0:00、8:00、16:00 促肾上腺皮质激素、皮质醇未见异常,节律正常。立卧位肾素、血管紧张素、醛固酮未见异常。促卵泡生成素5.09 IU/L(1.4~18 IU/L),促黄体生成素2.15 IU/L(1.5~34.6 IU/L),催乳素12.44 ng/mL(2.1~17.7 ng/mL),雌二醇24.12 pg/mL(0~40 pg/mL),孕酮0.25 ng/mL(0.28~1.22 ng/mL),睾酮108.15 ng/mL(241~827 ng/mL)。17α 羟孕酮、人绒毛膜促性腺激素、甲胎蛋白、血氨、乳酸无异常;糖化血红蛋白6.9%,胰岛细胞抗体、谷氨酸脱羧酶抗体、胰岛素抗体均阴性,口服葡萄糖耐量试验(OGTT)提示2 型糖尿病,见表1。
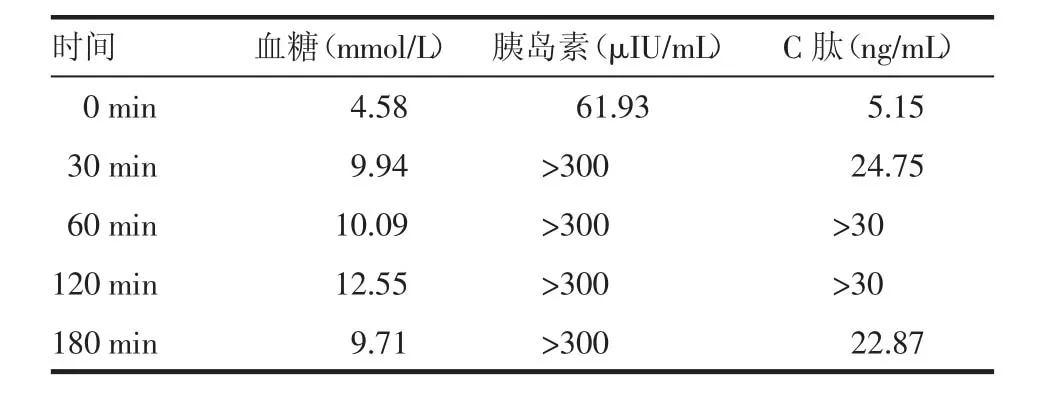
表1 患儿OGTT 试验结果
腹部超声示:肝大,下界剑突下23 mm,右肋下43 mm,轻度脂肪肝。泌尿系超声示:双侧肾上腺未见明显占位,输尿管、膀胱未见异常,右侧睾丸大小约2.9 cm×1.4 cm×1.9 cm,左侧睾丸大小约2.8 cm×1.2 cm×1.8 cm,双侧睾丸微小结石症,附睾、精索未见异常。心脏超声示:二尖瓣返流,三尖瓣轻度返流。
在获得知情同意后,抽取患儿及其父母静脉血各2 mL。全外显子捕获测序方法行基因检测发现患儿ALMS1 基因编码区外显子8 纯合子c4907-4910delTAAA,在蛋白序列中发现了移码突变,同时使蛋白的合成提前终止。其母亲鉴定为杂合c4907-4910 del TAAA,父亲正常。上述基因检测由北京德易东方转化医学研究中心有限公司完成(图2)。

图2 外显子8 的4907-4910 位点突变序列分析
目前该患儿就读于智障学校,口服二甲双胍以减轻胰岛素抵抗,加用保肝、降脂类药物,同时保护肾脏,减轻尿蛋白。佩戴眼镜调整视力。加强运动,调整饮食,积极改善生活方式以期提高生活质量。
2 讨论
ALMS 由ALMS1 基因突变引起ALSM 蛋白缺乏所致。该基因位于染色体2p13,包含23个外显子,编码4169个氨基酸。尽管该基因的功能尚未被详细阐明,但该蛋白广泛表达于多种组织如中枢神经系统、光感受器、内分泌系统及泌尿生殖系统纤毛细胞中心体和基部[4],在细胞内物质运输、细胞周期调控、维持纤毛细胞功能和结构稳定、信号通路调节、能量代谢平衡及细胞分化中发挥着重要作用[5]。由此ALMS 也被为是人类遗传疾病中与纤毛功能障碍有关的一种纤毛类疾病[6]。迄今,已报道316 种ALMS1 基因突变,包括点突变、缺失、插入及框架位移等[7]。大部分突变发生在ALMS1 基因外显子8、10和16,也有少数报道突变位于外显子9、11、12、15、18 和内含子17 等;有学者发现约超过50% 的ALMS1 突变位于第8 外显子,与其较大的基因编码序列(49%)相当[8]。位于外显子8、16 的突变所致临床表型多复杂,病情较严重,表现为心脏、眼、耳、肝脏、肾脏等多器官病变,且有糖类、脂类代谢异常[9]。同时遗传修饰、环境或感染暴露、随机事件,均可导致ALMS 发病年龄和严重程度发生极大的差异。
本病临床表现复杂,主要包括:(1)视网膜色素变性,视神经萎缩。(2)神经性耳聋。(3)糖尿病,胰岛素抵抗。(4)心脏扩大、心力衰竭、扩张型心肌病。(5)肥胖、黑棘皮症。(6)高尿酸血症。(7)高脂血症。(8)性腺功能低下。(9)肝功能异常,慢性肝炎,肝硬化。(10)肾脏纤维化,缓慢进展的肾功能不全。(11)中枢性尿崩症。(12)身材矮小、脊柱侧突。(13)高血压等[10]。临床表现根据不同年龄各异。婴儿期易出现视网膜变性,部分病例因扩张型心肌病而表现为充血性心力衰竭;儿童期表现为听力下降、肥胖、高胰岛素血症和2 型糖尿病;在青春期及成年期更多的以典型2 型糖尿病、高甘油三酯血症、首发扩张型心肌病等为主要症状,严重胰岛素抵抗所致的黑棘皮病,进行性肝、肺、肾功能障碍,多器官纤维化,男性性腺功能减退症等可能陆续出现[11]。同时可合并多种内分泌激素紊乱,包括甲状腺功能减退症,胰岛素样生长因子系统的改变,男性睾酮水平低和女性的高雄激素血症[12]。
本例患儿肥胖,伴黑棘皮症,视力障碍,辅助检查提示2 型糖尿病、高甘油三酯血症、高密度脂蛋白-胆固醇降低,肝功能损伤、蛋白尿、脂肪肝等,临床特征符合ALMS。同时该例患儿发生ALMS1 基因c.4907_4910delTAAA 纯合变异,位于第8 外显子上,为未报道过的新变异。序列分析显示该变异导致氨基酸出现移码突变(p.N1637Kfs*4),蛋白质合成提前终止,产生截断的蛋白。根据美国医学遗传学学会(American College of Medical Genetics,ACMG)的判定标准,该变异位点为致病性(pathogenic)变异,其证据包括:(1)非常强烈的证据(pathogenicvery strong,PVS1):该变异为移码突变,可能导致基因功能丧失。(2)中等证据(pathogenic-moderate,PM2):在正常人群数据库中的频率(allelefrequency)为0.0006(包括1000 Genomes,esp6500,gnomAD 和ExAC 数据库)。(3)支持性的证据(pathogenic-supporting,PP4):临床表型吻合。
目前尚无针对ALMS 的特异性和根治性治疗手段,多为对症处理,早期诊断和生活指导有助于未来生活质量提高。基因治疗和干细胞治疗是未来的研究方向,已有一些研究证实了基因治疗技术在恢复纤毛功能方面取得成功[13]。
猜你喜欢
中国现代医生(2022年19期)2022-11-04
电子科技大学学报(2022年5期)2022-10-29
中老年保健(2021年4期)2021-08-22
中国生殖健康(2020年4期)2021-01-18
心肺血管病杂志(2020年5期)2021-01-14
趣味(数学)(2020年4期)2020-07-27
支部建设(2020年15期)2020-07-08
肿瘤预防与治疗(2019年6期)2019-07-30
腹腔镜外科杂志(2016年12期)2016-06-01
百科知识(2015年18期)2015-09-10
