
钙调控参与糖尿病小鼠冠状动脉平滑肌收缩变化的机制
2020-11-18 07:39周梦园郑丹琳秦晓玥李穗敏邝素娟邓春玉
中国药理学通报 2020年11期
张 利,周梦园,郑丹琳,秦晓玥,李穗敏,曾 鹏,邝素娟,杨 慧,饶 芳,邓春玉,3,4
[1. 华南理工大学生物科学与工程学院,广东 广州 510006; 2. 广东省人民医院(广东省医学科学院)医学研究部临床药理重点实验室,广东 广州 510080;3. 华南理工大学医学院,广东 广州 510006;4. 广东省心血管病研究所心血管内科,广东 广州 510100]
糖尿病患者首次确诊时,多已伴有心血管并发症,临床上冠状动脉粥样硬化是其主要的致死原因[1]。糖尿病血管内皮功能障碍是其重要的病理生理特征,近年来大量研究表明,糖尿病的发生也伴随着血管平滑肌细胞(vascular smooth muscle cells,VSMCs)功能异常,并影响着许多重要动脉的收缩。已有文献报道,糖尿病大鼠主动脉和冠状动脉的血管反应性发生变化,糖尿病主动脉的收缩反应减弱,冠状动脉的舒张功能减弱,而收缩变化不明显[2-3];然而,糖尿病小鼠胸主动脉血管收缩反应却明显增强[4],提示不同种属和器官的糖尿病血管反应性变化存在差异,但糖尿病对小鼠冠状动脉收缩影响的相关文献报道甚少,有待进一步阐明。
胞内游离Ca2+在介导VSMCs收缩功能中起着重要作用[5]。VSMCs内Ca2+浓度变化受肌浆网内钙释放和胞外钙内流的影响,其中L-型钙通道和钙库操纵性钙(store-operated calcium,SOC)通道是外钙内流的主要途径[6-7]。已有研究表明,糖尿病使L-型钙通道和SOC通道功能发生改变[2-4],提示细胞内钙调控异常与糖尿病血管反应性变化密切相关。因此,本研究通过给予不同的血管收缩剂5-羟色胺(5-hydroxyl tryptamine,5-HT)和血栓素A2类似物(9,11-dideoxy-11α, 9α-epoxymethanoprostaglandin,U46619)及不同信号通路阻断剂硝苯地平(nifedipine)和毒胡萝卜素(thapsigargin,TG),观察糖尿病冠状动脉血管张力变化,探讨糖尿病对钙调控参与冠状动脉收缩反应性的差异,以研究糖尿病对小鼠冠状动脉收缩的影响。
1 材料
1.1 实验药物U46619、5-HT(091M5163V)、硝苯地平(57H1139)、链脲佐菌素(streptozotocin,STZ;031M1287V)、毒胡萝卜素(049M4032V)、咖啡因(Caffeine;71K1877)、EGTA(111K5411)均购于Sigma公司;其余试剂均为国产分析纯。Krebs Henseleit(K-H)溶液(mmol·L-1):NaCl 119、NaHCO325、MgCl2·6H2O 1、KCl 4.7、KH2PO41.2、CaCl22.5、D-glucose 11.1;高钾K-H溶液(mmol·L-1):KCl 60、NaCl 63.7、NaHCO325、MgCl2·6H2O 1、KH2PO41.2、CaCl22.5、D-glucose 11.1。Ca2+-free K-H溶液(mmol·L-1):NaCl 119、NaHCO325、MgCl2·6H2O 1、KCl 4.7、KH2PO41.2、D-glucose 11.1、EGTA 0.05。实验所用溶液配好后均通混合气(95% O2+5% CO2)充分饱和。
1.2 仪器620 M型小血管张力测定仪(丹麦DMT公司);PowerLab 8/30生物信号采集处理系统(澳大利亚AD公司);Stemi DV4 型体视显微镜(德国ZEISS公司);DK-8D 型电热恒温水槽(上海医用恒温设备厂);Advatage血糖仪和血糖试纸(Roche)。
1.3 实验动物SPF级,8~9周龄C57BL/6小鼠,♂,共30只,购于江苏集萃药康生物科技有限公司,许可证号:SCXK(苏)2018-0008,饲养于华南理工大学实验动物中心。本研究所有动物实验已通过广东省人民医院(广东省医学科学院)的动物实验伦理审查(NoGDRE201208a)。
2 方法
2.1 糖尿病小鼠模型的建立将C57BL/6小鼠随机分为模型组(model)和对照组(control),每组各15只。模型组和对照组通过腹腔注射分别注入等量STZ(50 mg·kg-1体质量)和溶剂(0.1mol·L-1柠檬酸-柠檬酸钠缓冲液,pH 4.2~4.5),连续5 d给药,同时给药前禁食12 h不禁水,给药后,再禁食2 h[4,8]。一周后使用罗氏血糖检测试纸和血糖仪,通过尾静脉采血法定期采集两组小鼠的血糖水平变化,之后每隔1个月测量1次血糖水平,测量2次,以最后1次测量的血糖水平为准,模型组小鼠血糖高于16 mmol·L-1视为造模成功。
2.2 冠状动脉环的制备血管环的制备同已发表文献[9-10],简而言之,通过CO2麻醉并处死小鼠后,快速取出心脏,放入4 ℃预冷的K-H(Krebs Henseleit)溶液中并借助体视显微镜去除冠状动脉周围的心肌组织,分离出冠状动脉并剪成长度为1.8~2.0 mm血管环;用两根直径为40 μm的不锈钢丝穿过血管环,并将其固定在张力测定仪浴槽内的两个钳夹上,同时钢丝要保持平行。在37 ℃温浴的K-H液中平衡30 min后,给予冠状动脉1.2 mN的基础张力,平衡60 min,期间每隔15 min换液1次,并使张力维持在1.2 mN。
2.3 药物对冠状动脉张力作用的检测血管张力反应性的检测同已发表文献[9, 11]。对血管功能性检测合格的血管,在浴液中加入单剂量5-HT(2 μmol·L-1),待血管收缩效应达到最大值后,用Ca2+-freeK-H液洗脱4次,给予1μmol·L-1硝苯地平和2 μmol·L-1TG孵育30 min,加入2.5 mmol·L-1CaCl2,观察冠脉的血管反应性变化;用Ca2+-freeK-H液洗脱并给予1 μmol·L-1硝苯地平孵育,加入20 mmol·L-1Caffeine,观察血管张力变化。
采用浓度梯度给药的方法,在浴液中分别加入各浓度梯度的血管收缩剂5-HT (0.001~10 μmol·L-1)和U46619 (0.000 1~1 μmol·L-1),观察血管收缩剂诱导血管张力的变化。当血管收缩效应达到最大值后,用K-H液洗脱4次,并加入1 μmol·L-1硝苯地平孵育30 min,重复上述5-HT和U46619浓度梯度给药,观察硝苯地平对血管收缩量效曲线的影响。当血管收缩效应达到最大值后,用Ca2+-free K-H液洗脱4次,并给予1 μmol·L-1硝苯地平孵育30 min,再次重复以上5-HT和U46619浓度梯度给药,观察在无钙条件下硝苯地平对血管收缩张力变化的影响。
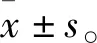
3 结果
3.1 糖尿病小鼠模型建立对照组与模型组小鼠分别注射等量溶剂和STZ,1周后,模型组小鼠出现明显的“多饮、多食、多尿和体质量减轻”症状,对照组与模型组小鼠的体质量分别为(18.70±3.06)g和(29.68±2.59)g(n=12,P<0.01);与对照组(5.1±1.70)mmol·L-1相比,模型组小鼠的血糖水平(25.84±4.48)mmol·L-1明显升高(n=12,P<0.01)。
3.2 小鼠冠状动脉收缩的钙调控机制高钾刺激模型组冠状动脉(2.84±0.40)mN(n=10)产生的收缩明显低于对照组(1.86±0.53)mN(n=12)(P<0.01)。在Ca2+-free K-H液中给予L-型钙通道阻断剂硝苯地平和钙泵阻断剂TG孵育后,加入2.5 mmol·L-1CaCl2,未引起冠脉收缩;给予20 mmol·L-1Caffeine引起血管急剧收缩。提示冠脉的平滑肌收缩主要由经L-型钙通道内流和肌浆网钙释放的钙离子介导,SOC通道不参与冠脉收缩,见Fig 1。
3.3 糖尿病小鼠冠状动脉对血管收缩剂5-HT和U46619的反应采用浓度梯度给药法分别加入血管收缩剂5-HT(0.001~10 μmol·L-1)和U46619(0.000 1~1 μmol·L-1),发现5-HT与U46619均可诱导小鼠冠状动脉呈明显的浓度依赖性收缩,模型组小鼠冠状动脉平滑肌收缩反应明显低于对照组。5-HT诱导模型组冠状动脉量效曲线的Emax以及pEC50(Emax:64.12±18.23,n=9;pEC50:6.42±0.25,n=9)均明显低于对照组(Emax:112.75±9.33,n=10;pEC50:7.12±0.20,n=10)(P<0.01)。U46619诱导模型组冠状动脉量效曲线的pEC50(7.17±0.34,n=9)明显低于对照组(7.78±0.33,n=8)(P<0.01),而Emax在两组间差异无显著性(P>0.05),见Fig 2。
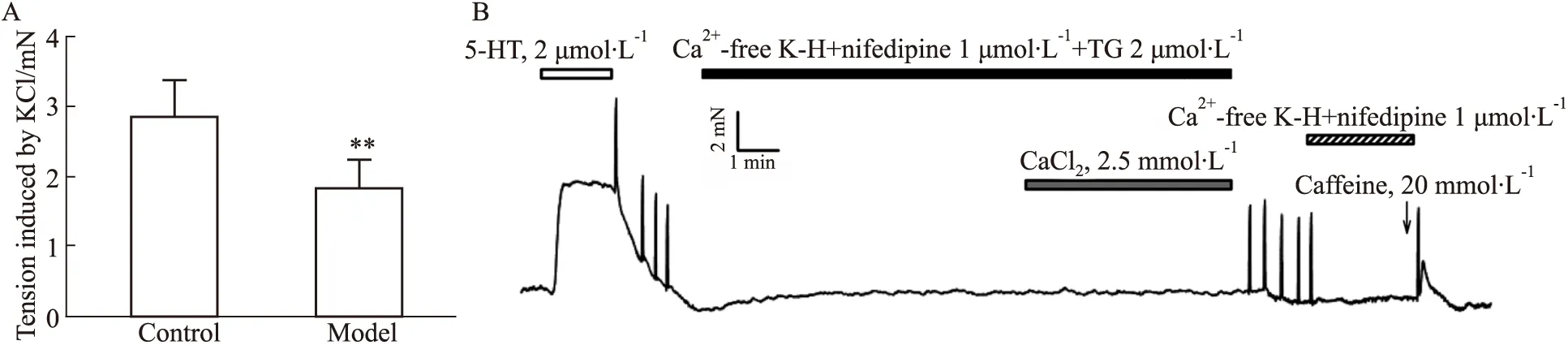
Fig 1 The mechanism of calcium handling involved in constriction of coronary artery in mice A: High potassium-evoked contraction was stronger in the coronary arterial rings of wild-type mice than of diabetic mice (n control=12, nmodel=10); B: Coronary arteries were washed in Ca2+-free buffer in the presence of nifedipine (1 μmol·L-1) and TG (2 μmol·L-1) for 30 min, and there was no response to CaCl2 at 2.5 mmol·L-1 in coronary arteries of mice, and caffeine at 20 mmol·L-1 significantly evoked constriction of coronary arteries in mice(n=3). **P<0.01 vs control.
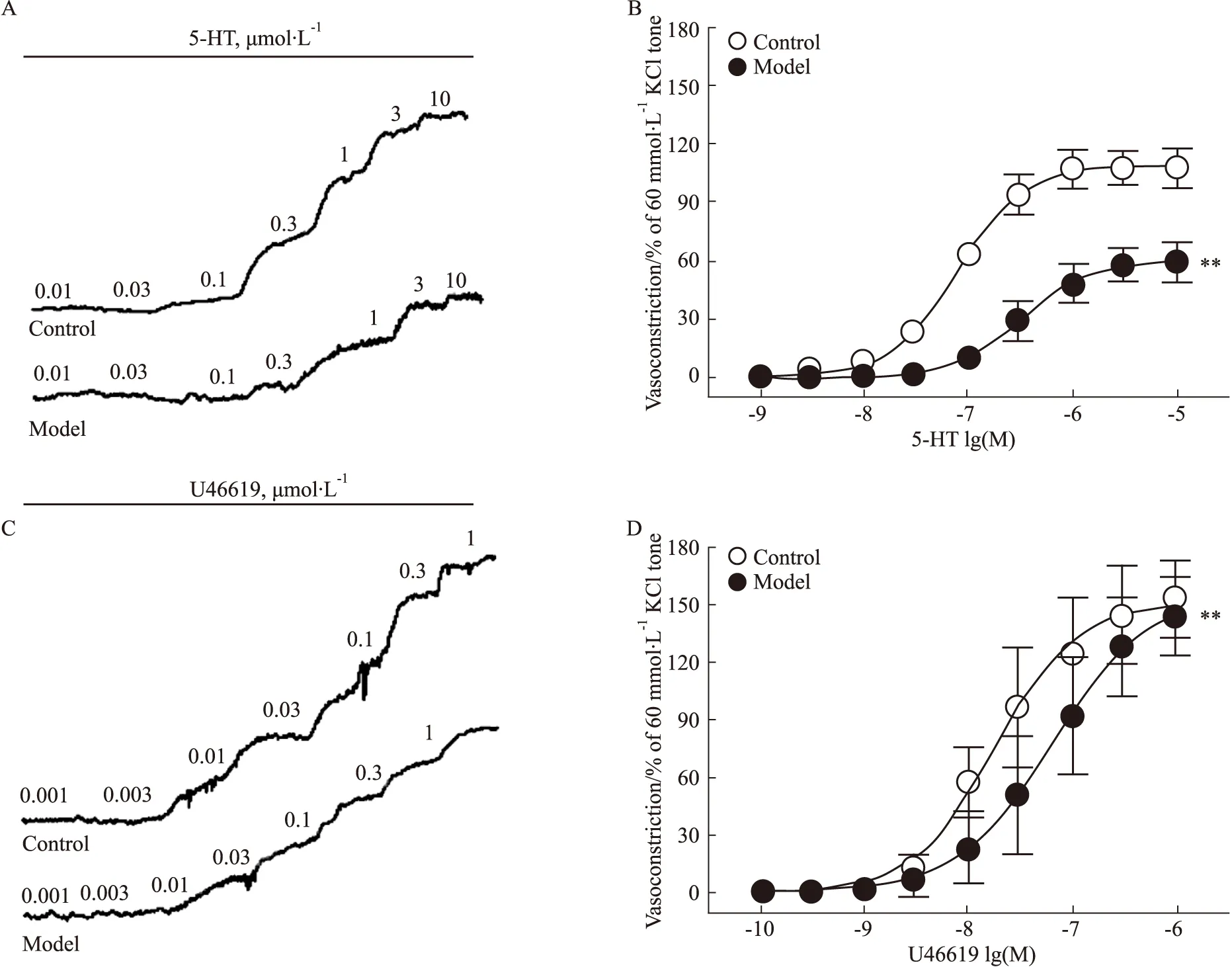
Fig 2 The concentration-dependent vasoconstriction induced by 5-HT and U46619 in coronary arterial rings of diabetic mice Representative recording of 5-HT(A)- and U46619 (C)-elicited concentration-dependent contraction in the coronary arterial rings of wild-type and diabetic mice. The concentration-dependent vasoconstriction induced by 5-HT (B) and U46619 (D) in the coronary arterial rings of wild-type and diabetic mice. 5-HT: ncontrol=10, nmodel=8; U46619: ncontrol=10, nmodel=9. **P<0.01 vs control.
3.4 硝苯地平对血管收缩剂诱导糖尿病小鼠冠状动脉收缩反应的影响加入硝苯地平(1 μmol·L-1)孵育后,采用浓度梯度给药法分别给予血管收缩剂5-HT(0.001~10 μmol·L-1)和U46619(0.000 1~1 μmol·L-1),记录冠状动脉张力变化。结果显示,硝苯地平可明显抑制血管收缩剂诱导冠状动脉的收缩反应;模型组冠状动脉收缩反应明显低于对照组,硝苯地平对模型组冠状动脉收缩反应的抑制幅度明显降低(P<0.01)。5-HT诱导模型组冠状动脉的量效曲线的Emax(6.41±6.73,n=9)明显低于对照组(15.75±4.15,n=10)(P<0.05),而两组间的pEC50差异无显著性(P>0.05)。在U46619诱导冠状动脉收缩反应,两组的Emax和pEC50差异均无统计学意义(P>0.05),见Fig 3。
3.5 肌浆网Ca2+释放在糖尿病小鼠冠状动脉收缩反应中的变化在Ca2+-free K-H溶液中加入硝苯地平(1 μmol·L-1)孵育后,采用浓度梯度给药法分别给予5-HT(0.001~10 μmol·L-1)和U46619(0.000 1~1 μmol·L-1),记录冠状动脉张力变化。结果显示,在细胞外液无Ca2+条件下,模型组小鼠冠状动脉的收缩量效曲线明显右移。在5-HT和U46619诱导冠状动脉收缩反应中,两组间的Emax和pEC50均无明显差异(P>0.05),见Fig 4。
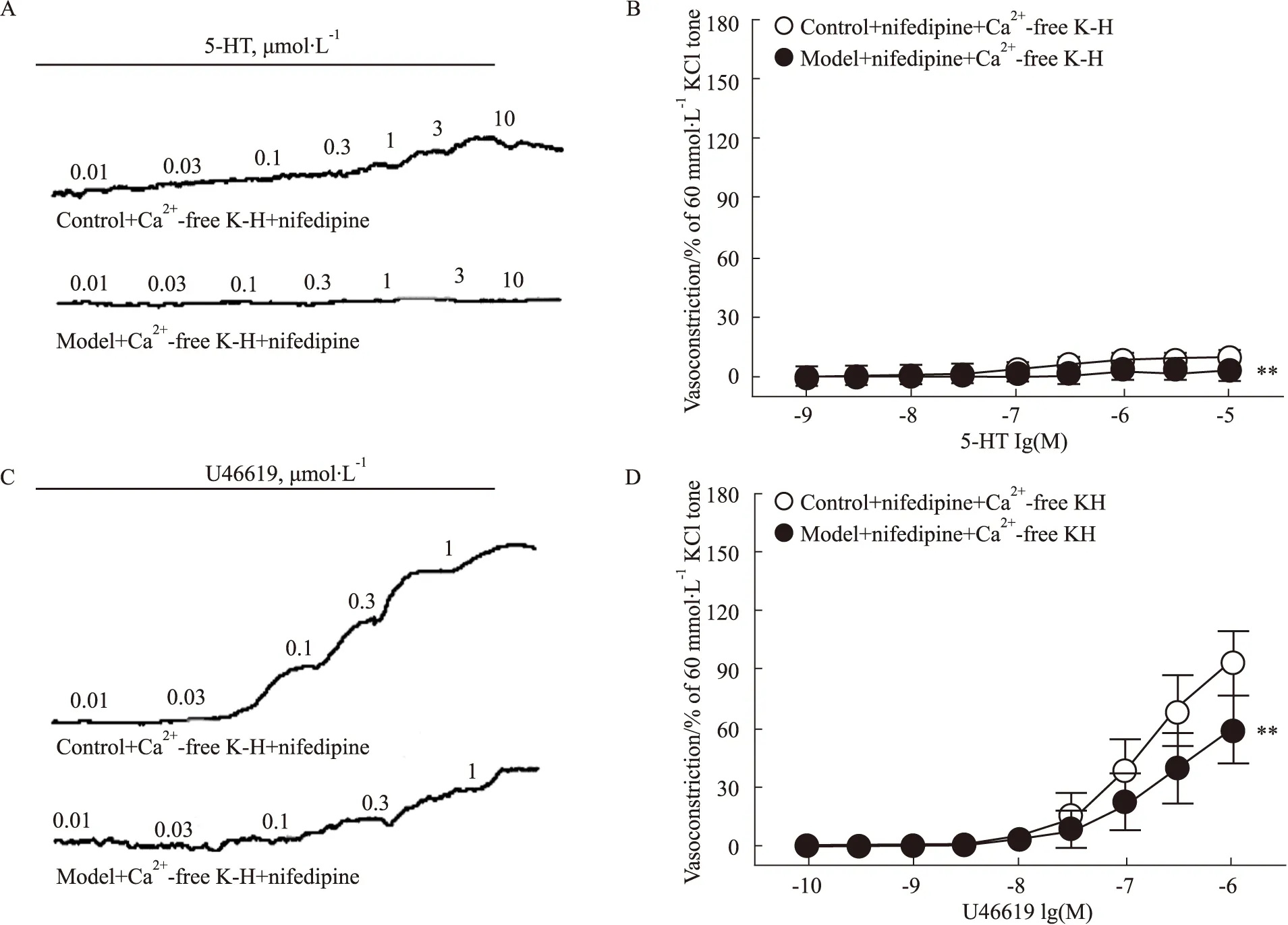
Fig 4 Effect of sarcoplasmic reticulum calcium on concentration-dependent vasoconstriction induced by 5-HT and U46619 in coronary arterial rings of diabetic mice Representative recording of 5-HT(A)- and U46619 (C)- elicited concentration-dependent contraction in the coronary arterial rings of wild-type and diabetic mice. In Ca2+-free buffer, effect of nifedipine (1 μmol·L-1) on concentration-dependent vasoconstriction induced by 5-HT (B) and U46619 (D) in the coronary arterial rings of wild-type and diabetic mice. 5-HT: ncontrol=8, nmodel=8; U46619: ncontrol=7, nmodel=9. **P<0.01 vs control.
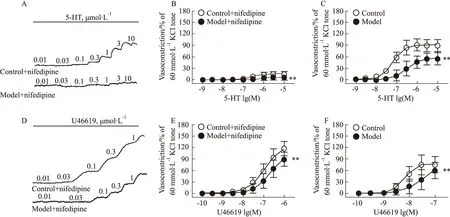
Fig 3 Effect of nifedipine on concentration-dependent vasoconstriction induced by 5-HT and U46619 in coronary arterial rings of diabetic mice Representative recording of 5-HT(A)- and U46619 (D)- elicited concentration-dependent contraction in the coronary arterial rings of wild-type and diabetic mice. The effect of nifedipine (1 μmol·L-1) on concentration-dependent vasoconstriction evoked by 5-HT (B) and U46619 (E) in the coronary arterial rings of wild-type and diabetic mice. The concentration-dependent vasoconstriction induced by 5-HT (C) and U46619 (F) inhibited by nifedipine (1 μmol·L-1) in the coronary arterial rings of wild-type and diabetic mice. 5-HT: ncontrol=11, nmodel=9; U46619: ncontrol=10, nmodel=9. **P<0.01 vs control.
4 讨论
VSMCs的收缩依赖于细胞内Ca2+浓度(intracellular calcium concentration,[Ca2+]i)的增加,[Ca2+]i的增加主要通过胞外钙内流和肌浆网内钙释放介导[5]。细胞外Ca2+内流途径主要包括与膜电位有关的L-型钙通道以及与钙库耗竭有关的SOC通道[6-7]。本课题组已研究报道2型糖尿病大鼠主动脉血管收缩反应性减弱与L型钙通道下调有关,SOC通道介导的收缩明显增强[2];但SOC通道不参与大鼠冠状动脉的收缩[12]。因此本实验通过不同血管收缩剂以及信号通路阻断剂,主要研究在有或无L-型钙通道介导下糖尿病小鼠冠脉收缩的差异性以及SOC通道和肌浆网钙释放功能的变化。
血管收缩剂5-HT和血栓素A2类似物U46619分别作用于VSMCs膜上的5-HT2A受体和血栓烷素(TP)受体[13-14],通过Gq蛋白激活磷脂酶C(PLC),促使磷脂酰肌醇二磷酸(PIP2)水解,生成三磷酸肌醇(IP3)和二酰基甘油(DAG),IP3与肌浆网上的IP3受体结合触发肌浆网内Ca2+释放,使膜去极化二次激活L-型钙通道,同时肌浆网内Ca2+耗竭可激活SOC通道,使细胞外钙内流,引起血管收缩;DAG可以通过作用于非选择性钙离子通道参与VSMCs收缩,另一方面还可以作用于蛋白激酶C(PKC),增加钙敏感性促进血管收缩[10, 15]。
为了阐明小鼠冠状动脉收缩的钙调控机制,采用高钾刺激血管引起收缩,提示L-型钙通道参与冠脉收缩;在细胞外液无Ca2+条件下加入硝苯地平阻断L-型钙通道介导的血管收缩以及肌浆网钙泵抑制剂TG进行孵育,发现加入CaCl2未引起冠脉收缩,而此时介导外钙内流参与血管收缩的途径为SOC通道,提示SOC通道不参与小鼠冠脉的收缩。同时实验中发现给予Caffeine后冠脉急剧收缩,而Caffeine通过诱导肌浆网Ca2+释放增加[Ca2+]i引起血管收缩,提示肌浆网钙释放参与小鼠冠脉收缩。
研究表明,糖尿病对血管平滑肌细胞的收缩功能产生影响[2-4],为了阐明糖尿病对小鼠冠状动脉血管张力的影响,本实验利用血管收缩剂5-HT和U46619诱导冠脉收缩,发现糖尿病小鼠冠状动脉收缩反应性较对照组明显减弱,提示糖尿病影响冠状动脉的收缩。实验中发现高钾刺激糖尿病小鼠冠状动脉引起的收缩反应明显减弱,而高钾使VSMCs膜去极化,使与膜电位有关的L-型钙通道开放,细胞外Ca2+流入胞内,引起血管收缩,提示糖尿病影响L-型钙通道介导的血管收缩。为了进一步验证糖尿病状态下钙调控机制的相关变化,采用L-型钙通道阻断剂硝苯地平进行孵育,加入收缩剂诱导血管收缩,发现硝苯地平对糖尿病组血管收缩反应的抑制幅度明显低于对照组,进一步说明糖尿病小鼠冠状动脉L型钙通道调控功能下调;并且糖尿病组对激动剂诱导的血管收缩明显低于对照组,提示糖尿病影响非L-型钙通道介导的血管收缩。随后,我们探讨糖尿病状态下肌浆网钙释放功能的变化,发现在含硝苯地平的无钙K-H溶液中,收缩剂诱导糖尿病组的血管反应性明显低于对照组,提示糖尿病小鼠肌浆网钙释放功能下调。
综上所述,本研究发现参与小鼠冠状动脉平滑肌收缩的钙调控机制主要包括L-型钙通道和肌浆网钙释放,SOC通道不参与其收缩;糖尿病小鼠冠状动脉对血管收缩剂的反应性明显减弱,与L-型钙通道和肌浆网钙释放功能下调有关。此外,Yokota等[16-17]研究表明,糖尿病患者对5-HT和血管紧张素II(Ang II)诱导血管收缩反应性增强与VSMCs膜上5-HT2A受体和AT1受体表达上调有关,糖尿病小鼠冠脉张力的变化是否也与VSMCs膜上相应受体的表达有关,本实验中未涉及,有待进一步研究。
猜你喜欢
中国药学药品知识仓库(2022年1期)2022-03-23
昆明医科大学学报(2021年12期)2021-12-30
昆明医科大学学报(2021年3期)2021-07-22
中国医药科学(2021年12期)2021-07-16
世界最新医学信息文摘(2021年12期)2021-06-09
世界最新医学信息文摘(2021年12期)2021-06-09
保健与生活(2020年24期)2020-12-23
中华养生保健(2020年10期)2020-12-03
中国药房(2019年22期)2019-10-20
中国医科大学学报(2017年10期)2017-10-12
