
当归腹痛宁滴丸质量标准提高
2020-11-18 07:24刘东升朱旭江姚世霞贺军权杨平荣
中国民族民间医药 2020年20期
朱 琳 刘东升 朱旭江 苏 蕊 姚世霞 贺军权 杨平荣
1.甘肃省药品检验研究院,甘肃 兰州 730070;2. 甘肃省中藏药检验检测技术工程实验室,甘肃 兰州 730070
当归腹痛宁滴丸具有解痉止痛的功效,主要用于妇女痛经、产后宫缩痛、感染性腹泻引起的急性腹痛、小儿秋季腹泻等的治疗。原标准收载于甘肃省药品标准1988年版。1998年收载入卫生部药品标准中药成方制剂第十七册,此标准沿用至今,此标准中收载性状、鉴别(当归)和检查项,标准较为简单,不足以控制当归腹痛宁滴丸的质量。所以有必要提高现行质量标准。当归腹痛宁滴丸是以当归挥发油为主要原料制成的滴丸剂,当归挥发油的主要成分为苯酞类,其中藁本内酯含量最高[1],藁本内酯有很强的解痉、平喘、镇定作用,临床上可用于治疗支气管炎和妇科疾病。研究显示,藁本内酯具有良好的改善微循环作用,可用于防治与微循环障碍有关的疾病[2-3]。与当归腹痛宁滴丸功能主治相吻合,因此当归腹痛宁滴丸的质量控制需定量测定藁本内酯的含量,确保药物的有效性。当归挥发油是当归经水蒸气蒸馏,再经二氯甲烷萃取而得到的,当归腹痛宁滴丸中有可能存在二氯甲烷溶剂残留,所以需要建立二氯甲烷溶剂残留检查项,确保药物的安全性。在验证原标准时还发现原标准中收载的当归薄层鉴别方法[4],由于展开系统极性太小且点样量过大,因此修改了当归薄层鉴别项。
1 材料
1.1 仪器 薄层色谱自动点样仪(CAMAG 半自动点样仪);薄层自动成像仪(CAMAG TLC成像仪);Agilent 7890A(CTC顶空进样器)气相色谱仪;Sartorius天平;电子天平(ME-204);Waters ALLiance e2695 型高效液相色谱仪;e2695 HPLC Pump、2998 DuaLλ Absorbance Detector 和Empower色谱工作站;资生堂CAPCELL PAK C18(4.6 mm×250 mm,5 μm)色谱柱,岛津Inertsil ODS-SP(4.6 mm×250 mm,5 μm)色谱柱和Welch Ultimate XB-C18(4.6 mm×250 mm,5 μm)色谱柱; KH-500DE超声波清洗器(上海超声波仪器厂)。
1.2 试药 当归对照药材(中国药品生物制品检定所提供,批号:120927-201014);藁本内酯(上海源叶生物科技有限公司提供,批号:Z23M7B15162,含量:98%);N,N-二甲基甲酰胺、二氯甲烷、甲醇与乙腈为色谱纯,水为超纯水,其余试剂均为分析纯;当归腹痛宁滴丸(兰州和盛堂制药有限公司提供,批号:20150401、20150402、20150403、20150406、20150407、20150408、20150409、20150701、20150702、20150703)。
2 方法与结果
2.1 薄层色谱鉴别 原标准中的当归薄层鉴别方法,由于展开系统极性太小,未将对照药材与样品中相应的斑点最大程度的展开(如图1A所示),所以调整展开系统的极性(如图1B所示)。另外,原标准中点样量过大,因此调整点样量。最终的方法改为:取本品20粒,置乳钵中,加乙醚研磨提取2次,每次10 mL,过滤,挥干乙醚,残渣加乙醇1 mL溶解,作为供试品溶液。另取当归对照药材0.5 g,加乙醚5 mL,振摇2 min,分取乙醚液,挥去乙醚,残渣加乙醇1 mL使溶解,作为对照药材溶液。照薄层色谱法(中国药典2015年版四部 通则0502)试验,吸取上述两种溶液各5 μL,分别点于同一硅胶G薄层板上,以正己烷-乙醚(7∶3)为展开剂,展开,取出,晾干,置紫外光灯(365 nm)下检视。供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的荧光主斑点。

A B A.原标准的薄层色谱图 B.展开剂极性与点样量修改后的薄层色谱图图1 薄层色谱图
2.2 二氯甲烷溶剂残留[5-7]
2.2.1 气相色谱条件 以6%氰丙基苯-94%二甲基硅氧烷为固定相的毛细管柱(DB-624,柱长为30 m,内径为0.32 mm,膜厚度为1.8 μm);柱温为程序升温:初始温度为30 ℃,保持3.5 min,以每分钟5 ℃的速率升温至100 ℃,再以每分钟50 ℃的速率升温至220 ℃,保持5 min(250 ℃ 后运行6 min);氢火焰离子化检测器检测,检测器温度250 ℃;进样口温度200 ℃;顶空进样,顶空瓶平衡温度为90 ℃,平衡时间30 min,进样体积300 μL。
2.2.2 溶液的制备
2.2.2.1 对照品溶液的制备 取二氯甲烷(色谱级)约30 mg,精密称定(31.5 mg),加N,N-二甲基甲酰胺稀释制成每l mL含0.3 mg的溶液,即得。
2.2.2.2 供试品溶液的制备 取重量差异项下的肠溶滴丸,研细,混匀,取本品约3.0 g,精密称定,置20 mL顶空瓶中,精密加人N,N-二甲基甲酰胺5 mL,密封,振摇使溶散,即得。
2.2.2.3 阴性样品溶液的制备 按制备工艺,提取当归药材的挥发油,配制不含二氯甲烷的阴性对照品,并按标准拟定的供试品溶液制备方法制成阴性对照液,即得。
2.2.3 专属性试验 精密吸取对照品溶液、供试品溶液、阴性样品溶液以及供试品溶液与对照品溶液的混合溶液,按“2.2.1”项下色谱条件进样,记录色谱图。结果表明,阴性样品色谱中,在与对照品色谱相应的出峰时间未出现相同的峰,且阴性制剂对所测组分无干扰。对照品溶液中,二氯甲烷的理论塔板数均大于100000,供试品溶液中二氯甲烷峰及其相邻色谱峰的色谱分离度均大于1.5,能够实现良好的基线分离。对照品溶液、供试品溶液及阴性样品溶液的色谱图如图2所示。
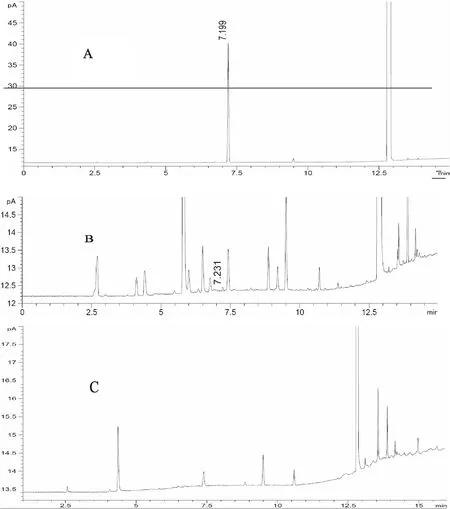
A.对照品;B.供试品;C.阴性样品 图2 二氯甲烷溶剂残留气相色谱图
2.2.4 线性关系的考察 取二氯甲烷(色谱级)约30 mg,精密称定(31.7 mg),加N,N-二甲基甲酰胺稀释制成每 mL含0.6 mg的溶液,摇匀,作为线性储备液备用;分别精密量取线性储备液体1.0、2.5、4.0、5.0、6.0、7.5 mL置10 mL量瓶中,N,N-二甲基甲酰胺定容,摇匀,分别测定分析,以峰面积为纵坐标,以样品浓度为横坐标,进行线性回归处理,得到二氯甲烷的回归方程为Y=94.818X+1.9965(R=0.999),结果表明,二氯甲烷在 0.0634~0.4755 mg范围内呈良好的线性关系。
2.2.5 精密度试验 精密吸取2.2.2项下的对照品溶液5 mL,置20 mL顶空瓶中,连续进样6次,记录峰面积,结果二氯甲烷的RSD为2.6%,表明仪器精密度良好。
2.2.6 检出限及定量限试验 将线性项下溶液图谱进行计算信噪比为10的为定量限,信噪比为3的为检出限之间。结果二氯甲烷的检出限为3.1500 μg/mL,定量限为12.6000 μg/mL。
2.2.7 稳定性试验 取同一供试品溶液,在0、2、4、8、12、16 h进行测定,记录峰面积,结果均未检出,表明供试品溶液在16 h内稳定。
2.2.8 加样回收率试验 精密量取2.2.4项下线性储备液适量,每三份一组,第一组加入线性储备液5.0 mL;第二组加入线性储备液10.0 mL;第三组加入线性储备液15.0 mL,分别置20 mL量瓶中用2.2.2项下的阴性样品溶液溶解并稀释至刻度,按供试品溶液制备方法制备,测定,计算回收率。二氯甲烷的平均加样回收率为97.7%,RSD为2.2%(n=9),本方法回收率良好。结果见表1。

表1 二氯甲烷加样回收率试验结果
2.2.9 样品测定 取10批样品(批号20150401、20150402、20150403、20150406、20150407、20150408、20150409、20150701、20150702、20150703),按“ 2.2.2.2”项下方法制备供试品溶液,按“2.2.1”项下色谱条件进行测定,记录测定结果。10批样品均未检出二氯甲烷溶剂残留。
2.3 藁本内酯的含量测定[8-10]
2.3.1 色谱条件 色谱柱选用资生堂CAPCELL PAK C18 (5μm,4.6 mm ×250 mm),以十八烷基硅烷键合硅胶为填充剂;以甲醇-乙腈-水 (15∶35∶50)为流动相,检测波长为327nm,柱温30 ℃,流速:1.0 mL/min,进样量:10 μL。
2.3.2 溶液的制备
2.3.2.1 供试品溶液的制备 取供试品,研细,取约0.5 g,精密称定,置50 mL量瓶中,加入甲醇约40 mL,超声提取15 min(功率500 W,频率40 kHz),放冷,用甲醇定容至刻度,摇匀,滤过,取续滤液,即得。
2.3.2.2 对照品溶液的制备 取藁本内酯对照品适量,精密称定,加甲醇制成每1 mL含藁本内酯50 μg,即得。
2.3.2.3 阴性样品溶液的制备 按处方比例及制备工艺,配制不含当归油的阴性样品,并按标准拟定的供试品溶液制备方法制成阴性对照液。
2.3.3 专属性试验 精密吸取对照品溶液、供试品溶液及阴性样品溶液,按“2.3.1”项下色谱条件进样,记录色谱图。结果表明,供试品色谱中,在与对照品色谱相应的出峰时间出相同的峰,且阴性样品溶液所测组分无干扰。供试品溶液中,藁本内酯的理论塔板数均大于10000,目标色谱峰及其相邻色谱峰的色谱分离度均大于1.5,能够实现良好的基线分离。对照品溶液、供试品溶液及阴性样品溶液的色谱图如图3所示。
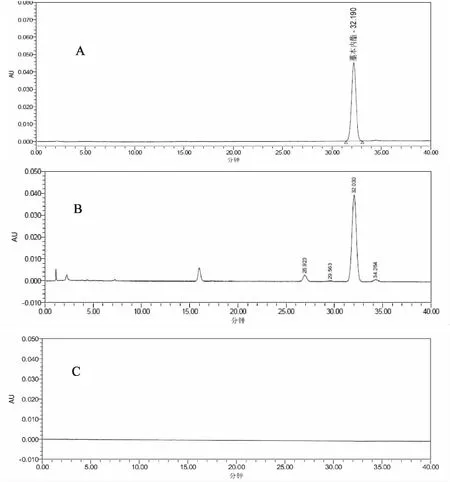
A.对照品;B.供试品;C.阴性样品 图3 当归腹痛宁滴丸液相色谱图
2.3.4 线性关系的考察 分别精密吸取“2.3.2.2”项下对照品溶液 2、5、10、15、20、30、40 μL注入高效液相色谱仪,在“2.3.1”条件下测定峰面积。以进样量(X, μg)为横坐标,峰面积(Y)为纵坐标,绘制标准曲线,得到藁本内酯回归方程为:Y=2760000X+3640,R=0.999999;线性范围为0.1064~2.1290 μg。
2.3.5 精密度试验 精密吸取同一对照品溶液10 μL,按“2.3.1”项下条件重复进样6次,记录峰面积。按峰面积计算,结果藁本内酯RSD为0.8%(n=6),表明仪器精密度良好。
2.3.6 重复性试验 取同一批号的当归腹痛宁滴丸样品6份(批号20150703),按“2.3.2.1”项下方法平行制备6份供试品溶液,各吸取10 μL进样,记录峰面积,结果藁本内酯含量的 RSD为1.8% (n=6),表明本方法重复性良好。
2.3.7 稳定性试验 取同一供试品(批号20150703)溶液,按“2.3.1”项下条件分别在0、4、12、24、48、96 h进行测定,记录峰面积,结果藁本内酯的RSD为1.2%(n=6),表明供试品溶液在96 h内稳定。
2.3.8 加样回收率试验 精密称取当归腹痛宁滴丸( 批号 20150703) 9 份,每份0.25 g,分为3 组。按供试品中待测化合物含量的50%,100% 和150% 分别加入对照品溶液(0.1330644 mg/mL、0.443548 mg/mL),按“2.3.2.1”项下方法制备供试品溶液,按“2.3.1”项下色谱条件进样测定,记录峰面积,计算藁本内酯的平均回收率为98.0%,RSD 为2.6%,结果见表2。

表2 藁本内酯加样回收率试验结果
2.3.9 样品测定 取10批样品(批号20150401、20150402、20150403、20150406、20150407、20150408、20150409、20150701、20150702、20150703),每批2份,按“ 2.3.2.1”项下方法制备供试品溶液,按“2.3.1”项下色谱条件进行测定,记录峰面积,测定含量。结果见表3。
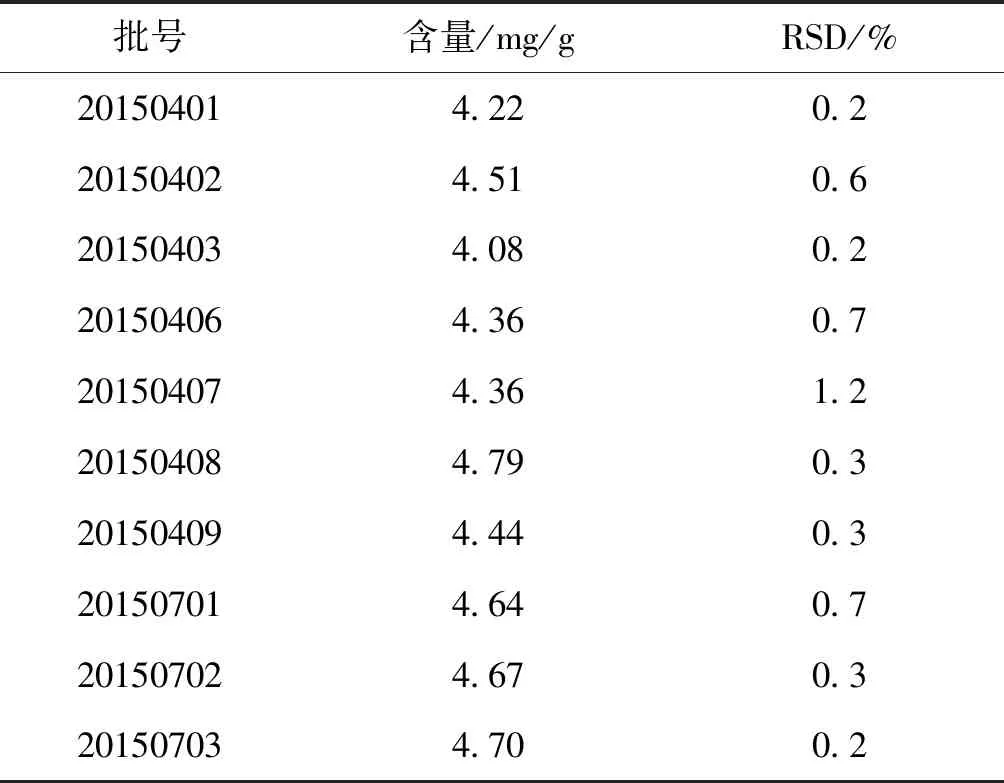
表3 样品测定结果 (n=10)
2.3.10 耐用性试验(不同色谱柱的影响) 为了考察该含量测定方法的耐用性,实验分别采用不同和品牌的色谱柱分别测定样品(批号:20150703)的同一供试品溶液,RSD为1.1%,说明不同色谱柱对其结果的影响较小。见表4。

表4 不同色谱柱测定结果
3 讨论
在藁本内酯的含量测定中,分别对以下四种流动相进行了考察:1.甲醇-水2.甲醇-1%醋酸溶液;3.乙腈-水;4.甲醇-乙腈-水。流动相1、2和3均不符合系统适用性要求,1和2不能使藁本内酯峰与其左侧峰的分离度达到要求,3不能使藁本内酯峰与其右侧峰的分离度达到要求,最后使用三相系统达到了色谱峰的很好分离,继续调整流动相比例,调整为甲醇-乙腈-水(15∶35∶50),保证主峰出峰时间尽量小的前提下,使主峰达到完全分离。对一批样品(20150409)分别采用甲醇、乙酸乙酯与三氯甲烷进行提取,三种溶剂的提取效率相似,但考虑到环保,所以采用甲醇为提取溶剂。
二氯甲烷溶剂残留检查项色谱条件的选择中:比较了不同色谱柱, 如HP-5、Rxi-17和DB-624等, 结果有的样品中有杂峰干扰, 有的二氯甲烷与相邻的峰分离, 最终选择DB-624(30 m×0.320 mm 1.80 μm) 为分析柱, 并对不同起始柱温和保持时间 (70 ℃ 7 min、50 ℃ 5 min和30 ℃ 3.5 min) 进行了比较 (后续升温速度和保持时间均相同), 结果在30℃ 3.5 min的起始温度条件下, 目标峰得到基线分离。依据中国药典2015年版规定,二氯甲烷限量为0.06%。实际样品中检测到的二氯甲烷残留量均远在药典规定的限量以下。方法学研究结果显示本法具有足够的灵敏度, 可检出样品中二氯甲烷的含量分别0.0008%。表明本法能有效控制当归腹痛宁滴丸原料药中残留溶剂量。
综上所述,本实验采用薄层色谱法和高效液相色谱法对当归腹痛宁滴丸分别进行了定性和定量鉴别,采用气相色谱法建立二氯甲烷溶剂残留的检查项。本方法专属性强,结果准确,可用于当归腹痛宁滴丸的质量控制,为临床安全科学用药提供了指导。
猜你喜欢
现代食品科技(2022年5期)2022-05-30
化学工程师(2022年3期)2022-04-19
今日农业(2021年2期)2021-11-27
牡丹江医学院学报(2021年5期)2021-10-28
西北药学杂志(2021年1期)2021-02-03
今日农业(2020年23期)2020-12-31
天津中医药(2020年5期)2020-06-01
科技与创新(2018年11期)2018-11-29
科技与创新(2015年20期)2015-10-29
科技与企业(2015年20期)2015-10-21
