
大鼠Klotho蛋白水平随血压升高而降低
2020-11-16 03:26王彦琛杨伟张玮罗丹苏显明
生物技术通讯 2020年4期
王彦琛,杨伟,张玮,罗丹,苏显明
1.咸阳市中心医院 老年病科,陕西 咸阳 712000;
2.西安交通大学第一附属医院 老年心血管内科,陕西 西安 710061
高血压是一种发病率高、可导致全身组织器官产生多种严重并发症的慢性疾病,不仅给患者自身带来病痛,而且给患者及家庭也带来严重的心理及经济负担,已成为全球健康问题[1-2]。目前高血压的发病机制尚未完全阐述清楚。研究发现,Klotho(kl)蛋白能够修复受损内皮细胞,通过内皮释放含一氧化氮(NO)的体液途径来保护心血管系统免受损伤,推测与高血压发生有一定的关系[3-4]。我们在建立高血压大鼠模型后,检测了血浆中kl蛋白和NO水平随血压水平变化的规律,以明确kl蛋白与高血压发生发展的关系。
1 材料与方法
1.1 材料
7周龄雄性SD大鼠(体质量200±10 g)由西安交通大学医学院实验动物中心提供,随机分为高血压组和对照组,每组24只。造模组给予N'-硝基-L-精氨酸98 mg/d连续灌胃[5],对照组给予等体积生理盐水灌胃1/d,共28 d。
大鼠肾小管上皮细胞、大鼠肾小管上皮细胞完全培养基购于普诺赛公司;N'-硝基-L-精氨酸购于凡科维公司;PBS购于上海双螺旋生物科技有限公司;kl蛋白、血栓素(TXA2)、内皮素(ET)酶联免疫法、NO一步法、丙二醛(MDA)TAB法、超氧化物歧化酶(SOD)WST-1法试剂盒均购自南京建成生物工程研究所;倒置相差显微镜为OLYMPUS公司产品;酶标仪购于上海赛默飞世尔科技有限公司;BP-300A全自动大小鼠无创血压测量系统购于成都秦盟软件有限公司。
1.2 样本采集与处理
实验开始前先分别用 0、100、200 μmol/L 的N'-硝基-L-精氨酸干预大鼠肾小管上皮细胞,24 h后Real-time PCR检测kl基因的表达变化。
分别于造模成功停药后第 7、14、21、28 d称重,测安静状态下大鼠尾动脉血压3次,取平均值,每组抽取6只体质量相近的大鼠,用10%水合氯醛按0.35 mL/100 g麻醉,取腹主动脉血EDTA抗凝,离心血浆,-20℃保存,用于检测kl蛋白、TXA2、ET、MDA、NO含量及SOD活力。采血后立即处死大鼠,快速取出胸主动脉,生理盐水冲洗,10%甲醛固定,常规石蜡切片,分别用于HE染色、免疫组化CD31标记。
1.3 PCR检测kl基因的表达
将肾小管上皮细胞传代、计数、冻存,细胞生长至密度80%左右进行加药处理,各组加入N'-硝基-L-精氨酸的浓度为 0、100、200 μmol/L,培养24 h后提供细胞进行荧光定量PCR检测。检测所需的kl及β-actin引物从万科生物科技公司订购,引物序列见表1。
1.4 血浆kl蛋白、TXA2、ET含量测定
采用ELISA试剂盒,酶标仪测定D450nm值,根据标准品吸光度及浓度绘制标准曲线,根据标准曲线计算样本浓度。
1.5 血浆NO、MDA含量及SOD活力测定
分别采用试剂盒测定血浆NO、MDA含量及SOD活力。
1.6 统计学分析
采用SPSS19.0软件进行统计学分析,实验数据以x±s表示,采用t检验比较2组大鼠实验初始的收缩压。采用析因设计方差分析干预后2组大鼠收缩压的变化。采用两因素多水平析因设计对2组大鼠的kl蛋白浓度,NO、ET、TXA2、MDA含量及SOD活力进行分析整理。
2 结果
2.1 动物模型的建立
实验开始前用不同浓度的N'-硝基-L-精氨酸干预大鼠肾小管上皮细胞,Real-time PCR检测,结果显示N'-硝基-L-精氨酸影响kl基因的表达,因此在干预药物停药1周后测定实验数据,避免造模药物影响实验数据的准确性。PCR产物扩增的特异性经扩增曲线及熔解曲线确定(图1),用2-△△Ct法计算mRNA表达水平(图2)。
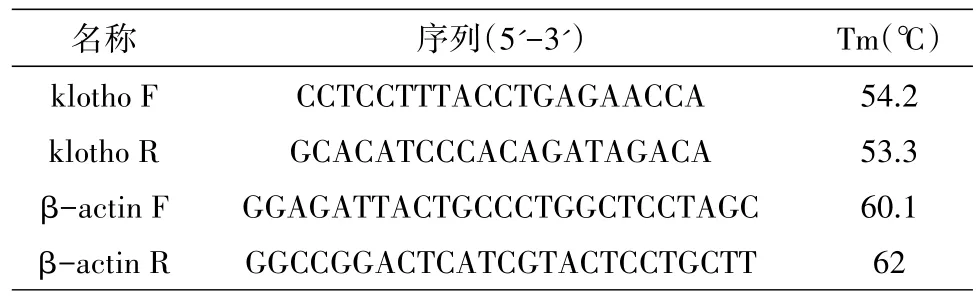
表1 引物及序列

表2 2组大鼠用药前收缩压比较(n=48)
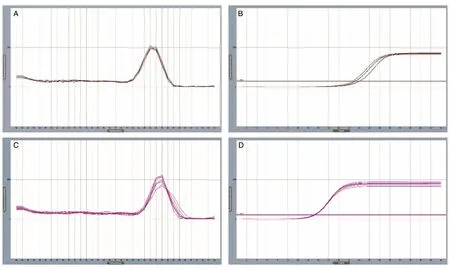
图1 kl基因(A)和β-actin(C)的熔解曲线及kl基因(B)和β-actin(D)的扩增曲线
2.2 血压水平结果
实验初始分别检测48只大鼠的收缩压,2组大鼠的收缩压差异无统计学意义(t=0.820,P=0.417)(表2)。经药物干预后,分别在停药后的第7、14、21、28 d测量2组大鼠的收缩压,结果见表3。采用析因设计方差分析2组大鼠的收缩压(表4)。在α=0.05水准上,校正模型检验F=171.841,P=0.000,模型具有统计学意义。在是否用药与时间的交互作用统计分析结果中,F=19.706,P=0.00,需要进一步阐释。在分析是否用药的单独效应结果中,F=1126.251,P=0.000,差异有统计学意义,即高血压组的收缩压与对照组的收缩压的影响有显著性差异,且高血压组的收缩压明显高于对照组。进一步分析7、12、21、28 d的收缩压变化情况,结果表明差异有统计学意义(F=21.064,P=0.000),而且随着时间的推移,血压不断升高。
2.3 各组 kl蛋白、NO、TXA2、ET、MDA 含量及SOD活性的变化
采用N'-硝基-L-精氨酸诱导大鼠高血压实验模型。为评估实验模型的有效性,实验中引入TXA2、ET、MDA含量及SOD活力检测。
对2组大鼠的kl蛋白、NO、ET、TXA2、MDA含量及SOD活力进行分析、整理。通过血压、干预后持续时间2个因素进行多水平析因设计,血压分对照组、高血压组2个水平,干预后持续时间分为7、14、21、28 d共4个水平,采用两因素多水平析因设计,共8种组合,每组6例,共48例。实验设计,kl蛋白、NO、ET、TXA2、MDA含量及SOD活力情况分别见表5、图3、图4。

图2 不同浓度N'-硝基-L-精氨酸干预大鼠肾小管上皮细胞kl表达情况

表3 2组大鼠用药后收缩压变化情况

表4 2组大鼠用药后收缩压变化的方差分析结果

图3 2组大鼠ET、TXA2、MDA含量及SOD活力随时间变化趋势
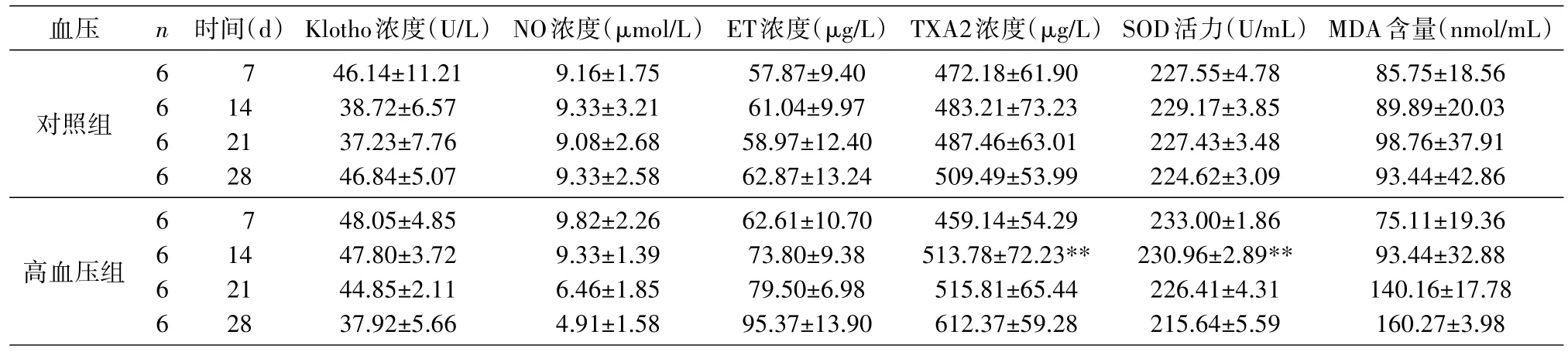
表5 2组大鼠用药后kl蛋白、NO、ET、TXA2、MDA含量及SOD活力情况描述
从析因设计方差分析结果(表6)可以看到,在α=0.05水准上,kl蛋白、NO、ET、TXA2、MDA含量及SOD活力的P值均<0.05,说明模型均有统计学意义。血压、时间之间的交互作用对kl蛋白、NO、ET、MDA含量及SOD活力的影响有统计学意义(均为P<0.05),但对TXA2含量的影响无统计学意义(P>0.05)。就血压的单独效应结果来看,单独血压对NO、ET、TXA2、MDA含量的影响有统计学意义(均为P<0.05),对kl蛋白浓度、SOD 活力的影响无统计学意义(P>0.05)。就时间的单独效应结果来看,单独时间对NO、ET、TXA2、MDA含量及SOD活力的影响有统计学意义(均为P<0.05),对kl蛋白浓度的影响无统计学意义(P>0.05)。

图4 2组大鼠kl蛋白、NO随时间变化趋势

表6 2组大鼠用药后kl蛋白、NO、ET、TXA2、MDA含量及SOD活力析因设计方差分析结果
2.4 血压变化与主动脉壁HE染色及CD31标记
HE染色结果显示,对照组大鼠主动脉管壁随时间变化,细胞核的大小、排列、管壁厚度未见明显的病理改变;高血压组大鼠随着高血压的发生,主动脉壁弹力纤维增生、排列紊乱,细胞核逐渐增大变形,中膜逐渐增厚(图5)。
CD31标记显示,对照组大鼠主动脉内皮细胞随时间无明显变化,边缘连续、光整,各时期无显著差别;模型组大鼠随着高血压的发生,内皮细胞受损,逐渐减少、脱落,部分完全缺失(图6)。
3 讨论
kl基因是Kuro等在1997年研究自发型高血压时发现的与衰老有关的新基因[6]。研究发现,人和大鼠的kl基因有83%的同源性,主要在肾脏和脑表达。该基因敲除鼠出生后4周龄左右出现动脉硬化和异位钙化,并随增龄而加重[7]。将kl杂合子鼠和野生型鼠同时放在高压环境下20 h后,杂合子鼠主动脉对乙酞胆碱松弛反应的有效剂量增高,小动脉对乙酞胆碱所引起的舒血管反应明显减弱[3],推断kl蛋白能够通过内皮释放NO产物的体液途径来保护心血管系统免受损伤。另有研究发现,kl蛋白可以减少过氧化氢所诱导的内皮细胞凋亡和衰老,并增强血管内皮细胞活性,降低 caspase-3、caspase-9 的活性[8]。缺乏 kl基因的小鼠,其骨髓、外周血中的内皮祖细胞(EPC)数量明显下降,而EPC的生理作用是当血管受损时可以迁移、黏附到受损部位,通过增殖、分化作用为内皮细胞修复受损的血管,因此考虑kl可能与EPC相互作用,参与血管损伤修复[9]。本课题组既往临床研究[10]了高血压组与非高血压组人群的kl蛋白与NO水平,发现高血压患者血清中kl蛋白随NO的增加而增加,kl蛋白与NO之间存在正相关,推断kl蛋白的降低可引起NO减少,而NO减少使NO/NOS系统功能低下,造成血管收缩、舒张不平衡,促进动脉粥样硬化的发生,最终导致高血压的发生。
本实验通过观察非高血压和高血压大鼠生长发育及疾病发生发展过程中kl蛋白、NO的变化,进一步推测kl与NO之间的关系。通过上述数据的分析(表6),我们可以观察到,仅血压的单独效应或者仅时间的单独效应对kl蛋白的影响无明显统计学意义,但血压及时间交互作用后对kl蛋白的影响有统计学意义,说明kl的表达变化在高血压发生发展进程中有时间累积效应,而非受短期血压波动影响。我们发现随着时间的延长、血压的逐渐升高,kl蛋白发生了规律性变化:kl总体呈逐渐下降趋势,但7~21 d高血压组大鼠kl蛋白水平明显高于对照组大鼠,21~28 d高血压大鼠kl蛋白水平低于对照组。高血压早期的病理生理变化是血管内皮损伤,而既往研究显示,kl蛋白在一定条件下可以与机体内的多种血管活性物质发生作用,使血管的舒张作用加强,抑制动脉硬化的发生,最终达到对血管内皮细胞的保护作用[11-12]。且该蛋白可以与VEGFR-2受体发生作用,保持内皮细胞的完整性[7]。因此推测在高血压初期,kl蛋白可能因为血压升高、反馈表达增加,发挥其保护血管内皮、降低血压的作用;而随着血压的稳定高水平,kl蛋白浓度显著降低,低于对照组水平,说明长期高血压可影响kl蛋白合成。因kl蛋白主要表达于肾脏远曲小管及脉络膜[13],远曲小管是离子转运和分泌的重要场所,其功能受醛固酮和抗利尿激素的调节,有研究发现RASS系统中血管紧张素Ⅱ可抑制kl mRNA表达,故在RASS系统对肾脏kl表达调节中,Ang应发挥着主导作用,因此高血压发生时,RASS系统激活,抑制kl基因的表达,kl蛋白最终减低,支持我们所观察到的kl蛋白表达水平变化趋势。
从上述数据可以看出,NO在血压、时间及其2组之间交互影响时差异均有统计学意义。我们发现2组大鼠的NO也存在显著差异:NO水平随血压升高及时间延长总体趋势逐渐降低,与血压呈负相关;但7~14 d高血压组大鼠NO水平明显高于对照组大鼠,14~28 d高血压组大鼠NO水平低于对照组(图4)。已知内皮细胞是血管中NO的主要来源,内皮细胞通过合成和分泌的NO来参与调节血管张力、血压和血管重塑等生理作用。高血压早期内皮细胞受损,NO产生减少。但是我们观察到在高血压发生的7~14 d NO水平高于对照组,与kl蛋白的波动趋势一致,结合上述kl蛋白升高的原因,我们考虑kl蛋白通过影响内皮细胞来影响体内NO的含量,共同调节血压水平。
另有研究表示[14],kl基因缺陷的小鼠在出生4周后即可出现动脉硬化,并随增龄而加重,表现为主动脉严重钙化,肌性中动脉中层钙化、内膜增厚,肾脏小动脉严重钙化等,这些血管变化与人类动脉硬化非常相似。把外源性正常kl cDNA转导给kl基因缺陷小鼠后,其动脉硬化程度可显著改善。本实验的病理切片也发现,随着高血压的发生,出现了血管管壁弹力纤维增厚、排列紊乱,弹力纤维板层结构破坏;免疫组化CD31标记明确显示内皮细胞不连续,逐渐变薄、缺失等内皮损伤改变,且在28 d时内皮缺失,这与我们观察到的21~28 d时kl蛋白及NO显著降低一致。
以上研究结果说明,在高血压发生发展中,RASS系统激活,kl基因表达减低,伴随kl蛋白减少,血管内皮受损、动脉硬化加重,NO进一步产生减少,血管收缩舒张功能失调,氧化应激加强,最终导致血压进一步升高。提示kl蛋白可能通过内皮细胞调节NO的途径参与血压调节过程,参与高血压的发生发展。
猜你喜欢
保健医苑(2020年6期)2020-07-09
中国眼镜科技杂志(2019年9期)2019-11-11
心肺血管病杂志(2019年1期)2019-04-22
天然产物研究与开发(2018年11期)2018-11-30
华人时刊(2018年23期)2018-11-18
江苏卫生保健(2018年11期)2018-02-13
解放军健康(2017年5期)2017-08-01
中国新闻周刊(2017年16期)2017-06-15
中国循证心血管医学杂志(2017年11期)2017-01-12
现代检验医学杂志(2016年4期)2016-11-15
