
基于生物信息学分析的肺腺癌诊断及预后相关基因筛选
2020-11-12 08:18刘少博
激光生物学报 2020年5期
刘少博,黄 波
(锦州医科大学附属第一医院胸外科,锦州 121000)
肺腺癌(lung adenocarcinoma,LUAD)属于非小细胞肺癌的一种,是世界上最常见的癌症之一,也是最常见的临床病理类型[1]。目前肺腺癌的发生率逐年增加,呈现出年轻化的倾向,疾病初期症状少,发病迅速,死亡率高且预后差,因此对肺腺癌的诊断及预后的诊断是非常重要的[2]。目前由于无法确定肺腺癌潜在的分子机制,其早期诊断以及预后治疗都是比较困难的,多数患者被诊断时已经处于晚期[3-4]。随着医疗技术和分子生物学技术的飞速发展,肺腺癌的治疗方式也逐渐向分子靶向治疗过渡[5]。肺腺癌早期诊断及预后的分子标志物对于其治疗有很大价值[6-8]。通过对疾病的发生发展及基因组水平的研究,寻找预后的生物标志物以及影响预后的因素,对恶性肿瘤的早期发现、诊断治疗及预后评估有突出作用[9]。随着基因组微阵列和高通量测序技术的进步,生物信息学分析为研究肿瘤的发生发展提供了有效方法,基因芯片和RNA测序的广泛应用也极大丰富了肿瘤相关的数据,通过在线数据库可获取大量的与肿瘤相关的数据[10]。本研究通过在线数据库及分析工具对多组肺腺癌组织与正常组织的数据集进行整合处理,利用生物信息学方法分析肺腺癌发生、发展及转移的可能机制,讨论肺腺癌诊断及预后可能潜在的生物标志物,为探讨肺腺癌预后相关基因的筛选提供理论依据。
1 材料与方法
1.1 芯片数据获取及数据处理
通过Gene Expression Omnibus(GEO)数据库(https://www.ncbi.nlm.nih.gov/)提取符合标准的基因表达谱数据(GSE63459、GSE27262、GSE75037)。筛选标准:1)标本为LUAD组织和对应的癌旁组织;2)每个芯片数据集都包含信使RNA(messenger RNA,mRNA)且数量不少于8对,本研究所选GSE63459数据集包含33个LUAD样本和32个癌旁样本,GSE27262含有25个LUAD样本和25个癌旁样本,GSE75037含有83个LUAD样本和83个癌旁样本。
1.2 差异基因的筛选
GEO2R(http://www.ncbi.nlm.nih.gov/geo/geo2r)是GEO中基于R的网络应用程序的一种交互式网络工具,可对 GEO数据库中的数据进行差异分析。运用GEO2R对芯片中差异表达基因(differentially expressed genes,DEGs)进行处理,对3组数据集利用t检验的方法,定义校正后P<0.010和|Log2FC|≥1有统计学意义。通过上述方法分别筛选出3组具有意义的数据集,然后利用在线分析平台维恩图(http://bioinformatics.psb.ugent.be/webtools/Venn)绘制差异基因的Venn图,获取3个数据集共同表达的上、下调的差异基因。
1.3 差异基因富集分析
使用生物信息注释数据库DAVID 在线分析平台(https://david.ncifcrf.gov/)对DEGs在基因本体(gene ontology,GO)中注释,包括分子生物学功能(molecular function,MF)、细胞学组分(cellular components,CC)和生物学过程(biological process,BP)的GO功能富集。利用京都基因与基因组百科全书(kyoto encyclopedia of genes and genomes,KEGG)进行通路分析,设定P<0.050为显著性基因富集[11]。对差异基因进行GO和KEGG分类分析是为了确定有哪些遗传功能和细胞信号通路的变化可能与差异基因有关。
1.4 PPI 网络构建与关键基因的筛选
STRING(search tool for the retrieval of interacting genes)是一个搜索已知蛋白质之间和预测蛋白质之间相互作用的数据库,并提供蛋白质-蛋白质互相作用(protein-protein interaction,PPI)的数据。用STRING数据库(http://string-db.org/)分析肺腺癌组织和正常肺组织 DEGs之间的 PPI关系,构造出PPI网络,综合得分(combined score)>0.4被认为有显著性差异。将分析的数据导入 Cytoscape(https://www.cytoscape.org/)软件后,基于k核(k-core)算法,利用插件MCODE(molecular complex detection)发掘肺腺癌蛋白-蛋白相互作用网络中不同功能的基因模块,筛选出MCODE score得分最高连接最显著的模块,筛选标准设定为:Degree Cutoff=2,Node Score Cutoff=0.2,k-core=2,Max Depth=100。随后运用DAVID在线分析平台对最显著模块中的基因进行GO和KEGG分析,使用 Cytoscape软件中 Cytohubba程序包分析 PPI 网络,并从 DEGs的PPI 网络中筛选出高度连通性排名前10的基因作为关键基因。Cytohubba 使用多种拓扑算法预测在既定网络中重要节点和子网间的关系。最后利用插件BINGO对关键基因的生物学过程进行分析,并通过热图来构建关键基因的层次聚类,用来区分肺腺癌和非癌样本。
1.5 关键基因的生存分析
Kaplan-Meier plotter是基于EGA(European Genomephenome Archive,https://ega.crg.eu)、TCGA(The Cancer Genome Atlas,https://tcgadata.nci.nih.gov/tcga)和GEO数据库评估大量基因对生存影响的常用网站工具。GEPIA(http://gepia.cancer-pku.cn/)是一个在线的基因表达谱动态数据分析数据库,用于分析癌症和正常组织之间的表达差异以及总生存率。首先,利用 Kaplan-Meier plotter验证10个关键基因与肺癌患者预后总生存率的关系,然后,通过GEPIA数据库发掘在mRNA表达水平上癌与癌旁的表达差异,并用UALCAN数据库(https://www.ualcan.com/)分析符合要求的关键基因与肺腺癌病理分期、淋巴结转移状态的关系。人类蛋白质图谱[human protein atlas(HPA),https://www.proteinatlas.org/]是一个基于免疫组织化学(immunohistochemistry,IHC)的正常组织、癌症和细胞系蛋白质表达图谱的数据库[12]。用免疫组化法从人类蛋白质图谱数据库中检测肺腺癌与正常组织之间生存相关基因的蛋白表达,明确基因在蛋白水平上是否存在差异表达。
2 结果与分析
2.1 筛选差异表达基因
通过对基因表达谱进行数据标准化处理之后,鉴定出3个数据集的差异表达基因,其中GSE75037中有3 413个,GSE27262中有2 272个,GSE63459中有407个。3个数据集之间的重叠包含共同差异基因355个,如韦恩图(图1a)所示,其中包括肺腺癌组织和非癌组织之间的273个下调基因和82个上调基因。
2.2 差异基因的GO及KEGG分析
通过DAVID在线分析平台对差异基因进行功能和途径富集分析,利用GO分析,将所有差异基因同时富集到MF、CC和BP这3种生物学关系中,富集分析结果显示:LUAD相关基因主要参与细胞外基质组织、细胞黏附、胶原分解代谢过程、血管生成和基因表达正调控等生物过程;差异基因的产物主要参与细胞外基质、胞外区域、胞外体、胶原三聚体、细胞外间隙等细胞组分,主要发挥调节金属内肽酶活性、肝素结合、糖胺聚糖的绑定、转化生长因子结合和调节受体活性等分子功能(表1)。通过KEGG通路分析,得到具有统计学意义的信号通路(P<0.050),即下调的DEGs主要在细胞黏附分子、过氧化物酶体增殖物激活型受体(peroxisome proliferator activated-receptors,PPARs)信号通路、转化生长因子-β(transforming growth factor-β,TGF-β)信号通路、调节干细胞多能性的信号通路、肿瘤坏死因子(tumor necrosis factor,TNF)信号通路中富集,而上调的DEGs主要富集在血小板激活、胞外基质-受体信号通路、黏着斑信号通路、磷脂酰肌醇-3-激酶(phosphatidy linositol-3-kinase and protein kinase,PI3K-Akt)信号通路等。
2.3 蛋白互助网络的构建及最显著模块的分析
利用Cytoscape的插件MCODE,根据MCODE模块中 MCODE score 降序排序,选择得分最显著的模块,具体见图1b,此模块涉及的基因功能分析也采用DAVID分析平台进行分析。GO分析结果表明,最显著模块中的基因主要参与细胞的有丝分裂、促进后期复杂分解等过程,参与细胞核、细胞质、纺锤体等细胞成分的组成;KEGG路径分析显示,基因的重要模块主要富集在卵母细胞减数分裂和细胞周期(表2)。之后,通过STRING数据库和Cytoscape软件对差异表达基因进行PPI网络的构建(图1c)。
2.4 关键基因的筛选及预后分析
通过PPI网络可见其中共有318个节点,最大连接度为89,最小为1(图1c)。利用插件Cytohubba以连接度排序,取前10位基因为关键基因,分别为IL6、MMP9、VWF、SPP1、PPARG、CCL2、PECAM1、TIMP1、COL1A1、CDH5。利用这些基因进行层次聚类,结果表明,关键基因可以区分肺腺癌样本和非癌样本(图2a),其生物学过程分析如图2b所示。随后,利用Kaplan-Meier曲线对关键基因进行单变量生存分析,结果得出10个关键基因的其中9个在总体生存率(overall survival,OS)上存在显著差异,6个显著的关键基因表达水平的升高表现出显著的OS下降,而其余3个关键基因表达水平的升高则表现出了OS上升(logrankP<0.01)。上述结果提示这些基因可以作为监测预后的指标。
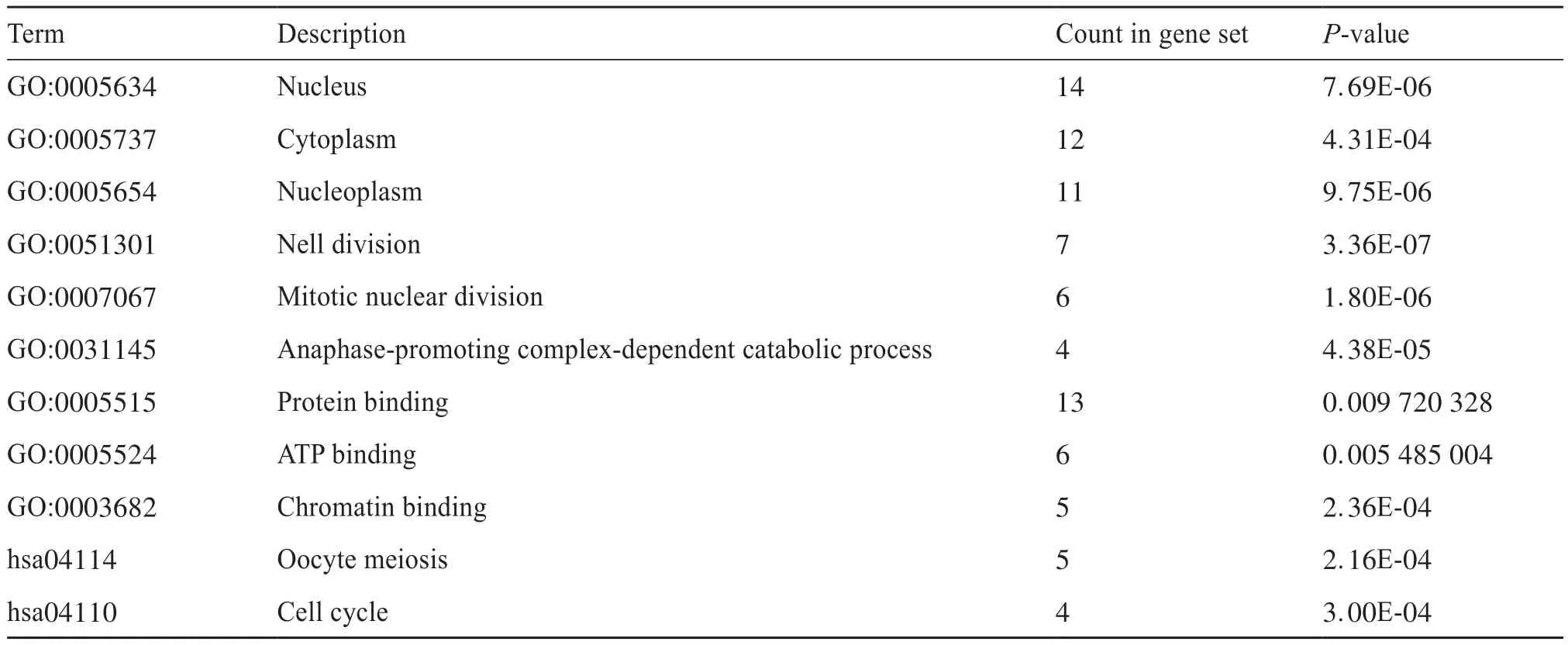
表2 最显著模块基因的GO和KEGG途径富集分析Tab.2 GO and KEGG pathway enrichment analysis of DEGs in the most significant module
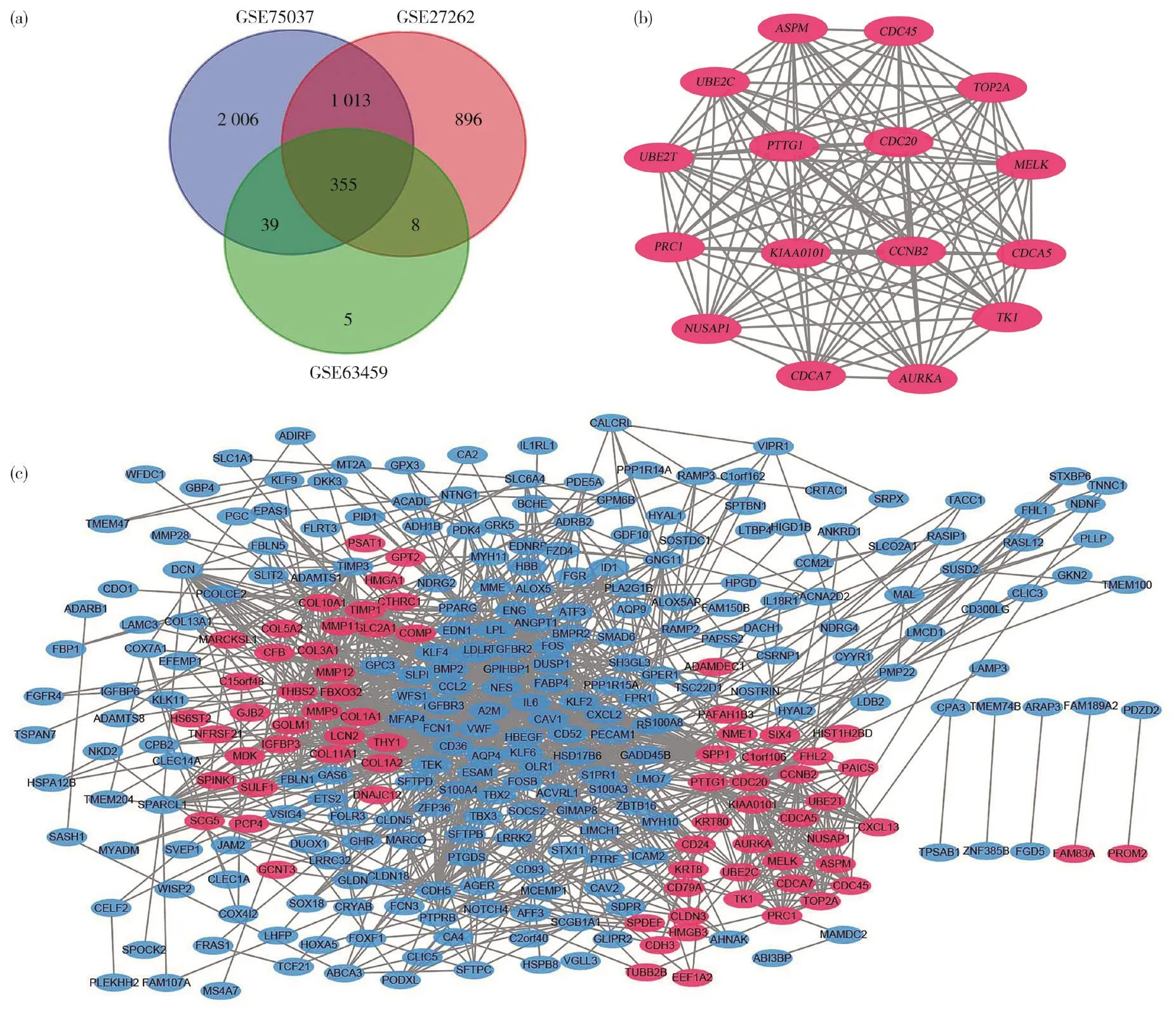
图1 差异基因的韦恩图、最重要的DEGs模块和蛋白互助网络图Fig.1 Venn diagram, the most significant module of DEGs and PPI network
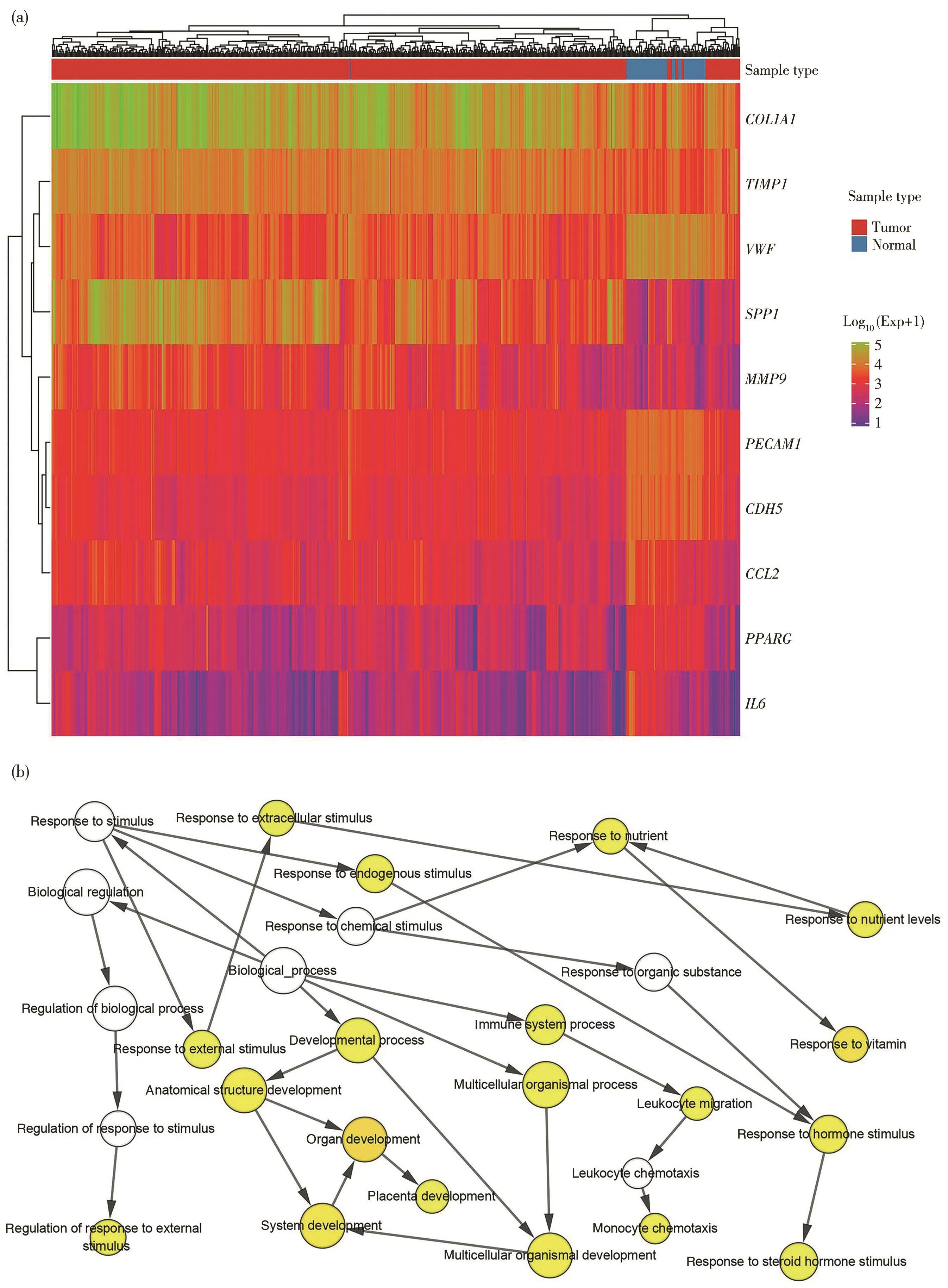
图2 关键基因的差异表达热图和生物学过程分析Fig.2 Differential expression thermogram and biological process analysis of key genes
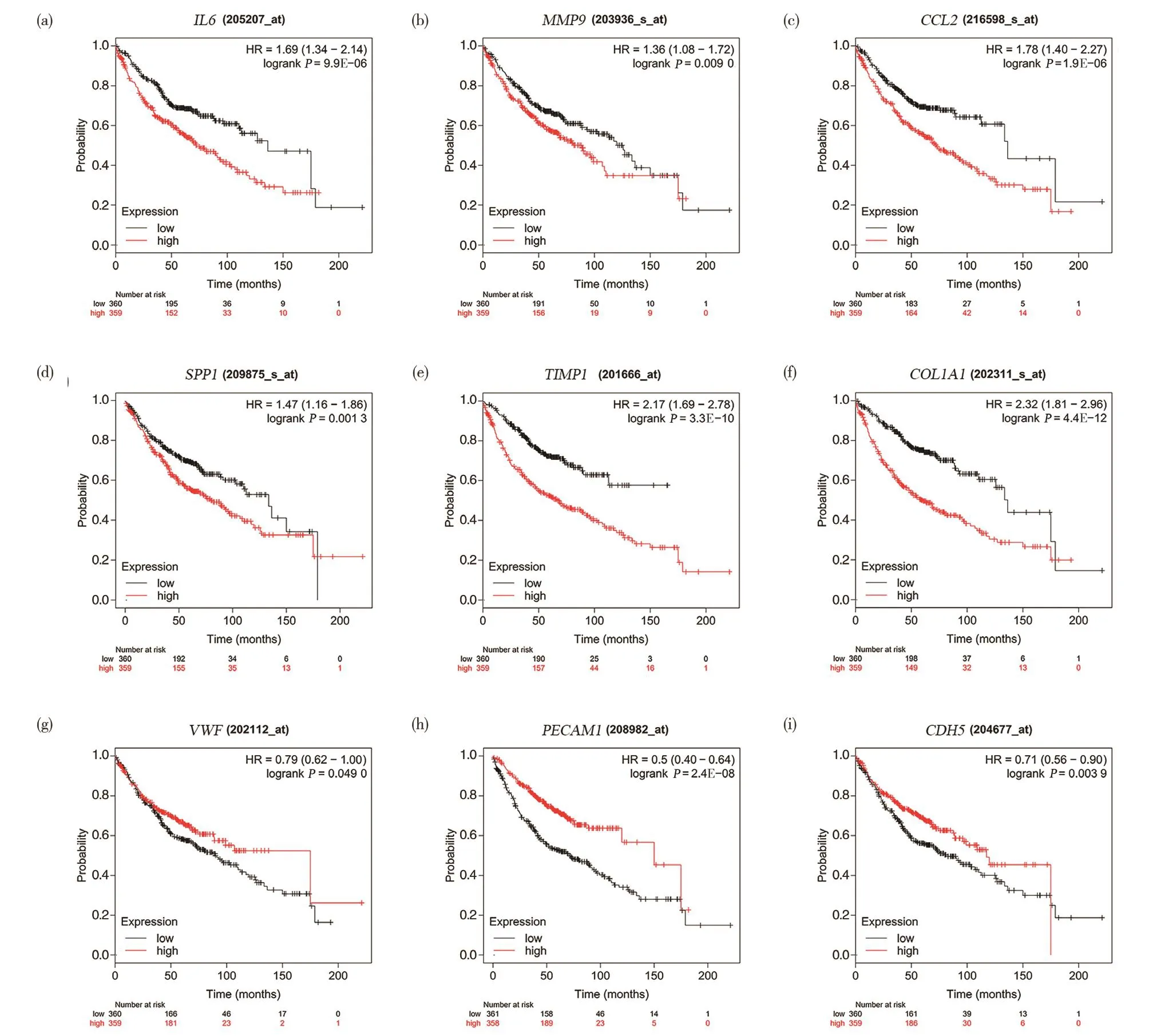
图3 9个核心基因总生存率分析Fig.3 Analysis of total survival rate of 9 core genes
2.5 关键基因的表达差异
通过Kaplan-Meier曲线分析得出IL6、MMP9、VWF、SPP1、CCL2、PECAM1、TIMP1、COL1A1、CDH59种关键基因对患者的总生存时间有着显著影响(P<0.050)。进一步利用GEPIA基于TCGA数据库分析上述基因在mRNA水平上肺腺癌与癌旁样本之间的表达差异,发现MMP9、SPP1、TIMP1、COL1A1在肺腺癌组织中明显高表达,而其他5种基因在肺腺癌组织中明显低表达。同时,利用UALCAN数据库分析癌组织中高表达的基因与肺腺癌病理分期、淋巴结转移状态的关系,具体结果见图4。另外从人类蛋白质图谱数据库获得了癌和癌旁组织中上述4种基因蛋白水平的免疫组织化学染色图像(图5),结果表明,LUAD组织中MMP9、SPP1、TIMP1和COL1A1蛋白水平高于正常组织,提示这些基因可能成为预后的分子标记物和治疗靶点。
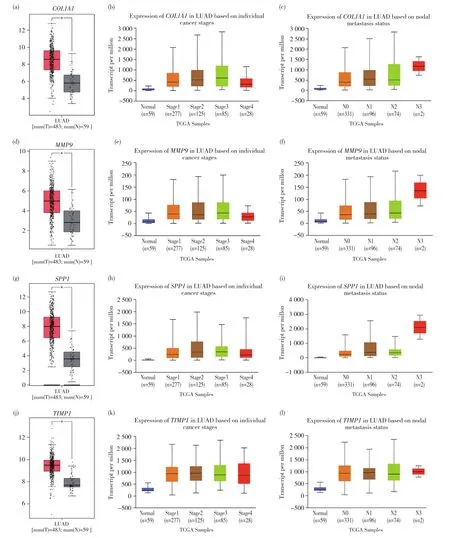
图4 COL1A1、MMP9、SPP1、TIMP1在LUAD肿瘤组织和邻近正常肺组织中的转录表达Fig.4 Expression of COL1A1, MMP9, SPP1 and TIMP1 in LUAD tumor tissue and adjacent normal lung tissue
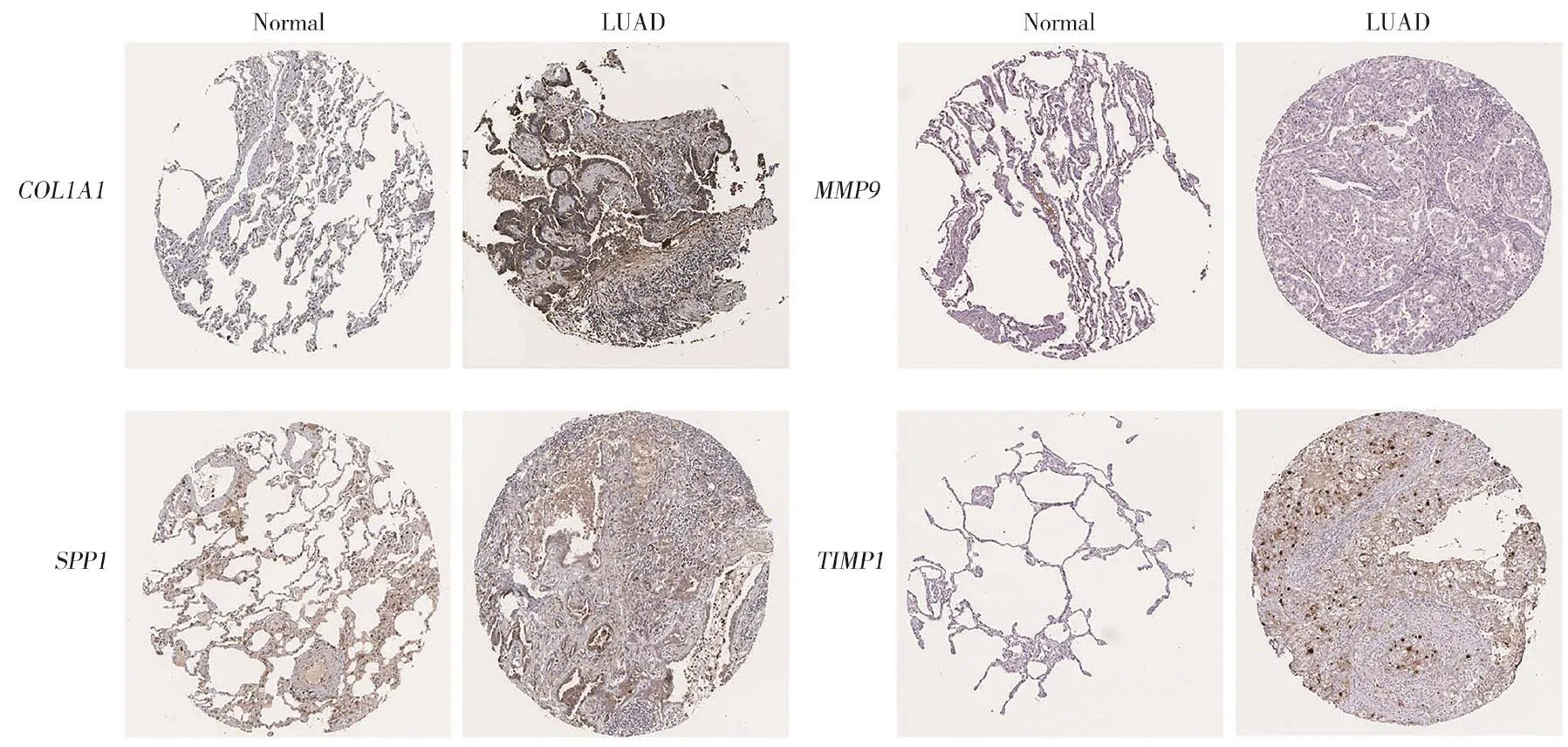
图5 COL1A1、MMP9、SPP1、TIMP1的LUAD组织和正常患者组织中蛋白表达的免疫组化染色Fig.5 Iimmunohistochemical staining of protein expression in LUAD tissues of COL1A1, MMP9, SPP1 and TIMP1 genes and normal tissues of patients
3 讨论
LUAD的发生发展是一个涉及多种基因和蛋白表达异常的复杂生理过程,由于恶性肿瘤的侵袭性强、易转移、生长快等特点,早期的诊断及预后治疗方法需要改进。微阵列技术和大规模序列技术的研究发展表明,基因对肿瘤的诊断和预后起着重要的作用[13-14]。因此,分析与LUAD侵袭和转移密切相关的基因尤为重要,可为早期诊断和预后治疗评估提供更多理论依据。为寻找LUAD早期诊断及预后的分子标志物从而达到肺腺癌患者的早期诊疗及预后的靶点治疗,本文对多种LUAD研究数据进行统一的方法处理,从基因和蛋白的层面剖析了LUAD的发展及转移的机制。
本研究对3组芯片数据进行挖掘,得到了355个差异基因,其中包括上调基因82个和下调基因273个。通过STRING数据库及Cytoscape软件对差异基因进一步筛选,得出10个关键基因,然后通过生存分析、基因及蛋白水平癌与癌旁的差异表达情况,得出SPP1、TIMP1、COL1A1、MMP94个基因,既往已有研究表明这4个基因可对癌症产生影响,本研究进一步从生物信息学角度验证了其可能对肺腺癌的诊断发展及预后有一定贡献。通过功能富集显示,上调差异基因主要富集在与细胞外基质(extracellular matrix,ECM)相关的细胞成分、生物学过程、分子功能以及信号通路中。而细胞外基质是肿瘤微观环境的重要组成部分,当肿瘤细胞脱落,便会黏附在细胞外基质,随着细胞外基质的分解向外侵袭和浸润。因此肿瘤细胞是否向别处转移和侵袭很大部分取决于细胞外基质的降解和细胞之间的黏附,这对肿瘤的预后有很大意义[15]。
组织金属蛋白酶抑制剂(tissue inhibitor of matrix metalloproteinases,TIMPs)是基质金属蛋白酶(matrix metalloproteinases,MMPs)的特异性抑制因子。TIMP1是由巨噬细胞和结缔组织细胞产生的一种糖蛋白,广泛存在于组织和体液中,可抑制包括MMP9在内的所有胶原[16],MMPS通过对细胞外基质的降解和突破基底膜的蛋白水解酶达到恶性肿瘤的侵袭和转移[17]。TIMP1既能抑制MMPS基质蛋白的水解,在一定程度上又可以激活MMPS从而抑制其降解作用[18]。另外有研究表明,TIMP1基因与大多数实体癌的侵袭和转移潜能密切相关,在前列腺癌[19]、结直肠癌[20]、肺癌[21]中的表达异常并可作为其侵袭转移的潜在的分子标志。另外,新生毛细血管的生成以及毛细血管的增生也可以促进肿瘤细胞的生长和扩散[22]。TIMP1还可与 MMP9形成1∶1的共价键复合物,抑制酶原活化和水解的作用,使细胞外基质的稳态处在动态平衡状态,而一旦两者的动态平衡被打破,便会促进肿瘤的侵袭和转移。利用GO及KEGG富集分析中可见,TIMP1和MMP9基因的表达在癌组织中上调,参与细胞黏附、血管生成,影响细胞外基质组成和金属内肽酶的活性,显著富集于胞外基质-受体和黏着斑信号通路,因此,TIMP1和MMP9可作为评估肺癌预后的独立指标和早期的分子标志物。
Ⅰ型胶原蛋白α1(typeⅠcollagen α1,COL1A1)是纤维胶原家族的主要成分,也是参与细胞外基质结构的组成的主要结构蛋白。研究发现COL1A1基因在胃癌[23-24]、乳腺癌[25]、食管癌[26]、肝癌[27]等多种恶性肿瘤的发生及转移中存在异常表达,并且可能与其预后相关。COL1A1的敲除缺失可以影响多种基因表达,从而抑制肿瘤细胞增殖,促进细胞凋亡[28]。另外,已有证据证明,COL1A1可诱导细胞间黏附复合物解聚和β-链蛋白的核转位从而促进胰腺癌细胞的扩散及增殖[29-30],并可通过TGF-β信号加快肺癌以及乳腺癌细胞中的上皮间质转化[31]。COL1A1基因在肺腺癌患者中使PI3K-Akt信号通路的许多组分比其他通路组分更容易被激活[32],胞外基质-受体和黏着斑信号通路也通过细胞黏附来影响肿瘤的预后与转移[33]。在本研究中,COL1A1基因在癌组织中上调且显著富集胞外基质-受体、TGF-β、PI3K-Akt、黏着斑信号通路之中,表明其有可能通过这4种通路参与LUAD发生发展的过程。
分泌性磷蛋白1(secretory phospho-protein1,SPP1)是一种富含趋化素样的基质磷酸糖蛋白,多存在于人体体液、肺、胃肠道、胰腺等多个器官,在多种肿瘤的发生和转移时都可见SPP1基因的高表达[34]。既往研究发现,RNA的干扰可以减少SPP1蛋白的表达,从而抑制肿瘤的生长,因此通过彻底敲除SPP1基因的方法可抑制肿瘤细胞生长[35-36]。据文献报道,在肺腺癌组织中尤其是浸润性腺癌中SPP1基因高度表达,其表达量与浸润转移程度相关[37]。特别对于相对早期的患者,SPP1可作为一种独立的具有预后意义的生物标志物[38]。另外SPP1还可通过NF-κB(nuclear factor kappa-B)依赖的信号转导通路促进基质金属蛋白酶的分泌,通过对细胞基底膜和细胞外基质的降解,使肿瘤细胞向远处转移从而造成更差的预后[39-40]。SPP1内含有的GRGDS(甘氨酸-精氨酸-甘氨酸-天冬氨酸-丝氨酸)序列可引起蛋白水解酶的激活从而降低细胞黏附作用,而且可通过抑制PI3KAkt信号通路抑制癌细胞的上皮间质转化[41]。在本研究中,SPP1基因在mRNA和蛋白水平在癌组织中表达均上调,而且多显著富集于胞外基质-受体、PI3KAkt、黏着斑信号通路之中,为肺腺癌的诊断预后治疗提供了方向。
综上所述,本研究对肺腺癌的芯片数据进行了挖掘及生物信息学分析,发现关键基因主要通过胞外基质-受体、黏着斑信号通路、TGF-β和PI3K-Akt信号通路等参与LUAD的发生发展过程。基因TIMP1、SPP1、COL1A1、MMP9可能是肺腺癌的潜在治疗靶基因,然而其具体的作用机制仍然需要更多的研究来证实。
猜你喜欢
感染、炎症、修复(2021年1期)2021-07-28
世界科学技术-中医药现代化(2020年2期)2020-07-25
心电与循环(2020年1期)2020-02-27
中成药(2018年7期)2018-08-04
江苏农业科学(2017年5期)2017-04-15
西南军医(2016年3期)2016-01-23
中国当代医药(2015年17期)2015-03-01
中华皮肤科杂志(2014年3期)2014-12-19
遵义医科大学学报(2014年1期)2014-08-09
湖北农业科学(2014年3期)2014-07-21
