
20批不同产地桂枝质量研究
2020-11-06 05:56于天颖周劲松马恩耀罗文英吴志坚党院霞孔箭
云南中医中药杂志 2020年10期
于天颖 周劲松 马恩耀 罗文英 吴志坚 党院霞 孔箭

摘要:目的 对20批不同产地桂枝的质量进行比较与分析。方法 采用薄层色谱法对桂皮醛及桂枝对照药材进行定性鉴别,同时依照2015年版《中国药典》(一部)“桂枝”项下进行显微、水分、灰分检查及浸出物含量测定。采用挥发油测定方法测定其含有的挥发油含量,采用高效液相色谱(HPLC)方法测定桂枝中含有的桂皮醛含量。并结合聚类分析及主成分分析方法对结果进行分析。结果 桂枝显微特征明显,薄层色谱斑点明显,不同产地的桂枝其水分含量范围在8.20%~10.83%;总灰分含量范围在0.56%~2.56%;醇溶性浸出物含量范围在6.14%~9.57%;挥发油含量范围在0.95%~1.66%;桂皮醛含量范围在1.01%~3.03%。结论 通过聚类分析得,可将20批不同产地的桂枝药材分为3类,结合主成分分析结果,其中广西平南的桂枝药材质量最好。
关键词:桂枝;桂皮醛;挥发油;聚类分析;主成分分析
中图分类号:R285.5
文献标志码:A
文章编号:1007-2349(2020)10-0057-06
桂枝,别名柳桂(Cinnamomum cassia Presl),是樟科植物肉桂的干燥嫩枝,主產于广西、广东、福建、云南等地[1]。味辛,温,入肺、心、膀胱经,是我国传统常用中药,药用历史悠久,在抗肿瘤、抗菌以及心脑血管保护、抗病毒等方面具有良好的药理活性[2-6]。
桂枝除含有桂皮醛、桂皮酸等还含有其他丰富的化学物质[7]。桂皮醛作为桂枝的基本化学成分,在去热、消痛、抗菌、消炎及其它方面效果明显[8]。由于桂枝的有效成分桂皮醛为挥发油,在储存过程中有可能因储存环境而损失,导致药效无法达到理想效果。所以测定其含有的挥发油含量具有极为重要的意义。
杨松等[9]运用高效液相色谱指纹图谱法等方法对不同产地和不同采收季节的桂枝样品,为桂枝质量标准研究打下基础。国内多为对桂枝合成药物如栝楼桂枝滴丸[10],栝楼桂枝颗粒[11],桂枝茯苓胶囊[12]等质量标准研究,对桂枝药材做详细质量标准研究少见。
已有文献研究中,对不同产地桂枝质量标准全方面的研究较少,因此本实验收集了广东、广西、安徽、四川以及福建等的20批桂枝药材,采用定性及定量分析方法对其质量评价,结合聚类分析及主成分分析比较不同产地的相关性,全面评价不同产地桂枝的质量,为桂枝药材的产地选择提供依据,同时为临床合理用药提供依据。
1 仪器与试药
1.1 仪器 BSA225S-CW分析天平(赛多利科学仪器(北京)有限公司),RHP-100型高速多功能粉碎机(浙江永康荣浩工贸有限公司),CX51RF型生物数码摄影显微镜(JAPAN OLYMPUS CORPORATION),79-1磁力加热搅拌器(常州澳华仪器有限公司),GZX-9070 MBE型数显鼓风干燥箱(上海博迅实习有限公司医疗设备厂),KSW型电阻炉温度控制器(余姚金电仪表有限公司),HH-6型数显恒温水浴锅(常州澳华仪器有限公司),HY-2调速多用振荡器(江苏金坛市环宇科学仪器厂),TC-15套式恒温器(海宁市新华医疗器械厂),UltiMate3000型高效液相色谱仪(美国戴安公司)。
1.2 试剂与试药 水合氯醛、甘油(AR,均购自天津市致远化学试剂有限公司)、乙醇、甲醇、石油醚、乙醚、三氯甲烷、乙酸乙酯(AR,均购自广州化学试剂厂)。乙腈(色谱纯,美国Fisher公司);水为纯水。
桂皮醛对照品(中国食品药品检定研究院,批号:110710-201418,含量以98.9%计),桂枝对照药材(中国食品药品检定研究院,批号:21191-201605)。
1.3 样品 桂枝药材共收集了20批,其批号和产地见下表1。经高明教授鉴别确认为樟科植物桂枝。
2 方法
2.1 显微鉴别 照《中国药典》2015版四步通则2001 显微鉴别法检测。
2.2 薄层鉴别 (1)取本品粉末0.5 g,加乙醇10mL,密塞,浸泡20 min,时时振摇,滤过,取滤液作为供试品溶液。另取桂皮醛对照品,加乙醇制成每1mL含1 μl的溶液,作为对照品溶液。照薄层色谱法(通则0502)试验,吸取供试品溶液10 μl~15μl、对照品溶液2μl,分别点于同一硅胶G薄层板上,以石油醚(60℃~90℃)-乙酸乙酯(17:3)为展开剂,展开,取出,晾干,喷以二硝基苯肼乙醇试液。供试品色谱中,在与对照品色谱相应的位置上,显相同的橙红色斑点。(2)取本品粉末2 g,加乙醚10mL,浸泡30 min,时时振摇,滤过,滤液挥干,残渣加三氯甲烷1mL使溶解,作为供试品溶液。另取桂枝对照药材2 g,同法制成对照药材溶液。照薄层色谱法(通则0502)试验,吸取上述两种溶液各15μl,分别点于同一硅胶G薄层板上,使成条状,以石油醚(60~90℃)-乙酸乙酯(17:3)为展开剂,展开,取出,晾干,喷以香草醛硫酸试液,在105℃加热至斑点显色清晰。供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的斑点。
2.3 水分 照《中国药典》2015版四步通则0832 水分测定法之第四法测定。
2.4 总灰分 照《中国药典》2015版四步通则2302 灰分测定法测定。
2.5 浸出物 照《中国药典》2015年版四部通则2201 浸出物测定法之醇溶性浸出物测定法项下的热浸法测定,用95%乙醇作溶剂。
2.6 含量测定 照《中国药典》2015年版四部通则2204 挥发油测定法测定。
2.7 桂皮醛含量测定
2.7.1 色谱条件 色谱柱:Agilent C18(250 mm×4.6 mm,5 μm);流动相:乙腈-水(32:68,v/v);流速:1 mL·min-1;柱温:30 ℃;检测波长:290 nm;进样量:10 μL。
2.7.2 对照品溶液的制备 准确称取桂皮醛对照品约20 μg,置2 mL容量瓶中,加入适量的甲醇使溶解并稀释至刻度,振荡均匀,用0.45微孔滤膜过滤,滤液即为对照品溶液(每1 mL含桂皮醛10 μg)。
2.7.3 供试品溶液的制备 准确称取过四号筛桂枝粉末约0.5 g,置锥形瓶中,严封,准确量取甲醇25 mL,称定重量,使用超声波清洗器超声提取30 min,冷却后称定重量,滴加甲醇至超声提取前的重量,振荡均匀倒入带有滤纸的干净漏斗中滤过,准确吸取续滤液1 mL倒入规格为25 mL的容量瓶中,加甲醇把续滤液稀释至刻度,振荡均匀,用0.45微孔滤膜过滤,滤液即为供试品溶液。
2.7.4 方法学考察
2.7.4.1 精密度试验 取“2.7.2”项下的对照品溶液,按“2.7.1”项下色谱条件进样6次,测定其峰面积并计算。
2.7.4.2 重复性试验 取批号为YPA3J00001的桂枝样品3份,准确称量,按“2.7.3”项下办法配制供试品溶液,测定并计算其峰面积。
2.7.4.3 稳定性试验 取批号为YPA3J00001的桂枝样品0.5 g,准确称量,按“2.7.3”项下方法制备供试品溶液,并于0、2、4、6、8 h按“儀器与方法2.7.1”项下色谱条件进样,测定并计算其含量。
2.7.4.4 加样回收率试验 取批号为YPA3J00001的桂枝样品0.5 g,精密称量6份,计算样品中桂皮醛含量。再分别加入桂皮醛对照品溶液1 mL,并按“2.7.3”项下方法制备成供试品溶液。测定并计算桂皮醛含量,计算回收率。
2.8 统计学方法 本实验运用IBM SPSS Statistics 25软件对各指标数据进行主成分分析和聚类分析。
3 结果与分析
3.1 显微鉴别 显微镜下观察,桂枝粉末有明显的韧皮纤维、石细胞、木栓细胞、木纤维及油细胞明显。由可以看出韧皮纤维多成束存在;石细胞壁厚,类椭圆形;木栓细胞多角形,含红棕色物;木纤维条状;油细胞椭圆形;导管为具缘纹孔导管。如图1所示。
桂枝药材横切面能见明显的表皮细胞、油细胞及石细胞,形成层明显,髓部发达。如图2所示。
3.2 薄层鉴别 如图3所示。在与对照品色谱相应的位置上,各产地药材显相同颜色的荧光斑点。
如图4所示,在与对照药材色谱相应的位置上,各产地药材显相同颜色的荧光斑点。
3.3 水分、浸出物、总灰分及含量测定 对20批不同产地桂枝进行水分检查,对20批样品进行水分含量测定,其水分含量在8.26%~10.83%之间,其灰分含量在0.56%~2.56%之间,其浸出物含量在6.14%~9.57%之间,其挥发油含量在0.95%~1.66%之间,其桂皮醛含量在1.01%~3.03%之间,结果如表3所示。20批桂枝供试品及其对照品高效液相色谱图如图5~6所示。
3.4 方法学考察
3.4.1 精密度试验 根据“2.7.4.1项”下测得峰面积结果为:3.6074、3.5942、3.6088、3.6122、3.5953、3.5933。测定桂皮醛峰面积RSD为1.0%。表明含量测定的样品溶液进样的精密度良好。
3.4.2 重复性试验 根据“2.7.4.2项”下测得峰面积结果为:5.8462、5.8493、5.8541。得到桂皮醛峰面积RSD为0.4%。表明该方法的重复性良好。
3.4.3 稳定性试验 根据“2.7.4.3项”下计算得含量为:2.29%、2.28%、2.29%。得到桂皮醛含量RSD为0.5%。表明样品在8 h内性质保持稳定。
3.4.4 加样回收率试验 根据“2.7.4.4项”下计算加样回收率。结果如表4。结果表明该方法准确度良好。
3.5 主成分分析 运用IBM SPSS Statistics 25软件,对所测结果进行主成分分析,首先得出其特征值和贡献率,结果如下。
由表5可得,主成分1的贡献率最高,为75.565%,主成分2的贡献率为20.790%。主成分1的特征值大于1,表明主成分1基本可以包含20批不同产地的桂枝药材所检测指标的大部分信息,因此用这1个主成分代替原来4个指标,用来评价20批不同产地桂枝药材的质量水平。
由表6可得,主成分1中各成分的贡献均为正值。浸出物、挥发油和桂皮醛的指标差异不大,且贡献最大。
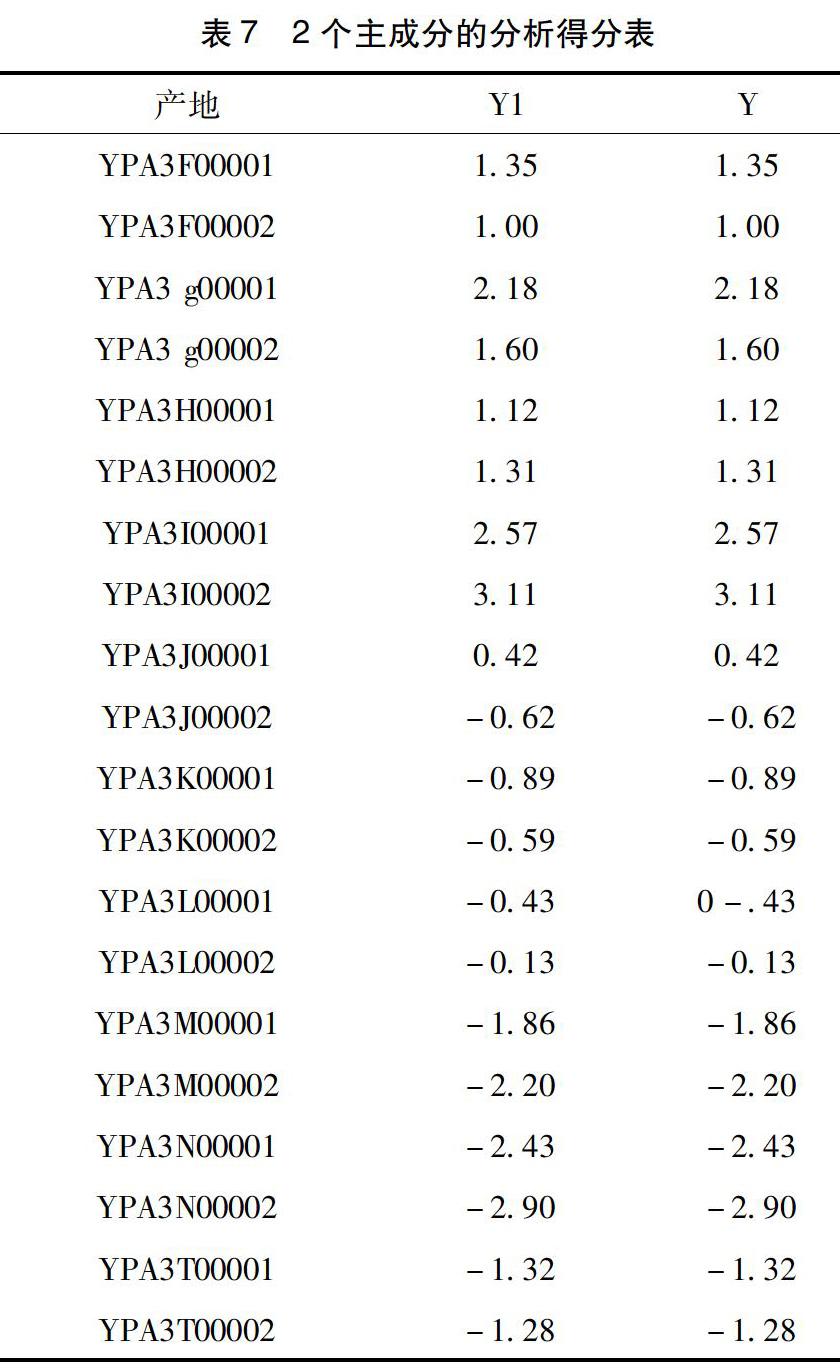
由表7可得,Y1为主成分1的分析得分值,Y为综合评分值。在主成分1中,负评分和正评分的桂枝药材产地基本各占一半,除广西桂平外,其他差异不大。而广西平南的主成分1最高,说明其桂枝药材的主成分1占有绝对优势。且由表10得,主成分1对总体的贡献率更大。最终得出结论:广西平南的桂枝药材质量最好,安徽合肥的评分最低,质量在20批桂枝药材中质量最次。
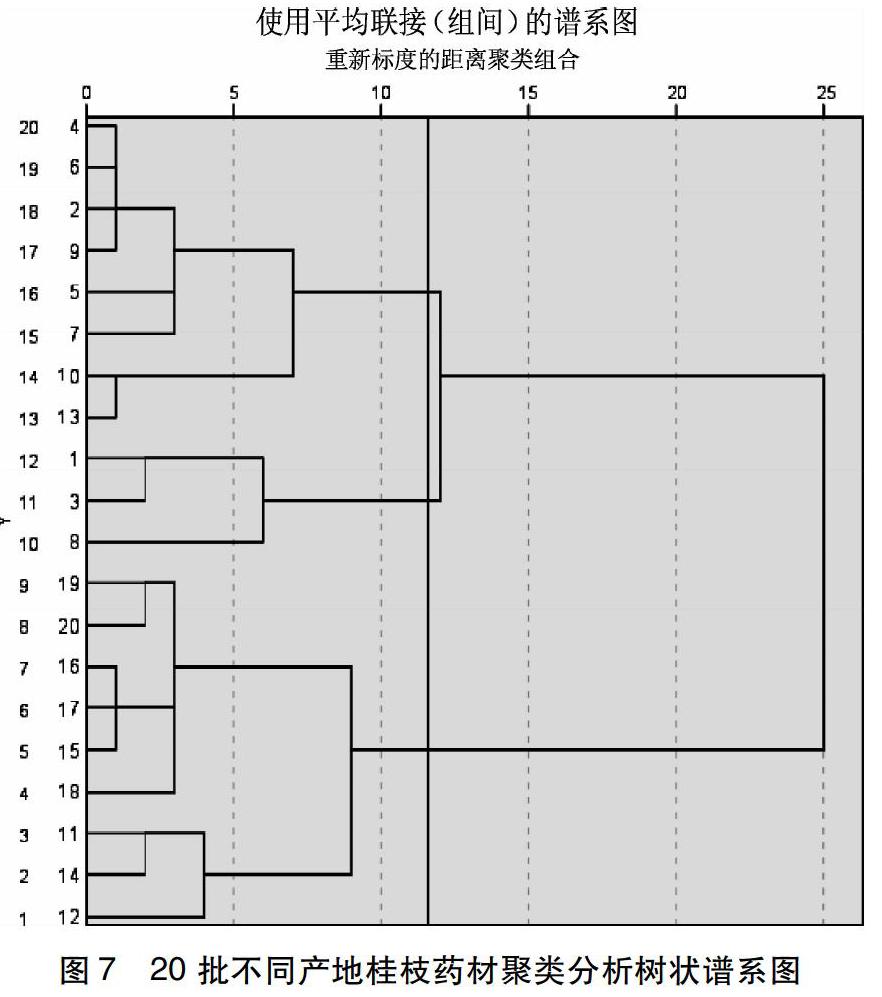
3.6 聚类分析结果 运用IBM SPSS Statistics 25软件,得到聚类分析树状谱系图,由图7可得,将20批不同产地的桂枝药材分为3类,交点以左即为各类的主成成分。其中1-9为第一类,10-12为第二类,13-20为第三类。
4 讨论
目前已报道文献中对桂枝药材的质量评价较为单一,更多的是根据药典对其含量测定条件的优化。选取单一指标并不能很好的评价桂枝药材质量。本实验增加样品批数,选取20批不同产地桂枝药材,对其进行定性及定量等全方面考察,以此来考察不同产地桂枝药材的质量,同时为桂枝药材产地的选择提供很好的依据。
对20批不同产地桂枝药材进行水分含量测定可知,含水量在8.26%~10.83%之间,均符合药典规定[13]。灰分反映药材中泥灰分土和砂石的量,以及生理灰分的量,20批不同产地桂枝药材的灰分在0.56%~2.56%之间,均也符合药典规定。桂枝药材在储存和运输中挥发油很容易损失,因此挥发油含量可以很好的评价桂枝药材的质量,对20批不同产地桂枝药材进行挥发油测定可知,含量在0.95%~1.66%之间,以广西平南所含挥发油含量最高。HPLC测定可知,其桂皮醛含量在1.01%~3.03%之间,均符合药典规定。采用聚类分析将20批产地桂枝药材分为3类,其广西平南的桂枝药材质量最好。
本实验对20批不同产地桂枝药材进行全面质量评价,确定不同产地桂枝药材质量优劣,不同产地桂枝药材含量差异较大,在聚类分析中,未发现含量与地域相关性,本实验为不同产地桂枝药材的选择提供了理论依据。
参考文献:
[1]国家中医药管理局《中华本草》编委会.中华本草(3卷)[M].上海:上海科学技术出版社,1999:42-45.
[2]LIANG M T,YANG C H,LI S T,et al..Antibacterial and antioxidant properties of Ramulus Cinnamomi using supercritical CO2 extraction[J].Eur Food Res Technol,2008,227(5):1387-1396.
[3]黄敬群,王四旺,罗晓星,等.桂皮醛对裸鼠人胃癌细胞移植瘤生长及凋亡的影响[J].解放军药学学报,2006,22(5):343-346.
[4]Verspohl E J,Bauer K,Neddermann E.Antidiabetic effect of Cinnamomum cassia and Cinnamomum zeylanicum in vivo and in vitro[J].Phytother Res,2005,19(3):203-206.
[5]蔣时红,李澜,吴耀松,等.桂枝茯苓丸抑制人乳腺癌细胞MCF-7增殖机制[J].中国实验方剂学杂志,2018,24(15):132-136.
[6]王瑞敏,侯懿,豆艳艳.桂枝茯苓丸和参麦散加减对宫颈癌患者术后放化疗的近期疗效观察[J].中国实验方剂学杂志,2014,20(6):187-191.
[7]朱伶俐,艾志福,徐丽,等.桂枝化学成分的分离鉴定[J].中国实验方剂学杂志,2019,2(23):1-5.
[8]乜红磊.指纹图谱结合多成分定量用于参枝苓口服液的质量评价研究[D].济南:山东大学,2017.
[9]杨松,鲁曼华,毕开顺.桂枝HPLC指纹图谱的研究[J].解放军药学学报,2005,21(3):217-219.
[10]李煌,马黄璜,乔丽菲,等.栝楼桂枝滴丸质量标准及体外溶出度研究[J].中国中医药信息杂志,2017,24(9):80-83.
[11]孙承韬,张玉琴,许文,等.栝楼桂枝颗粒质量标准研究[J].江西中医药,2017,48(8):58-62.
[12]丁玥,曹泽彧,柯志鹏,等.桂枝茯苓胶囊质量标准提升对制剂主要药效影响的研究[J].中国中药杂志,2015,40(19):3786-3793.
[13]国家药典委员会.中华人民共和国药典,一部[S].北京:中国医药科技出版社,2015.
猜你喜欢
中国药房(2019年12期)2019-10-20
小小说月刊(2018年5期)2018-05-18
大经贸(2016年9期)2016-11-16
中国市场(2016年33期)2016-10-18
科技视界(2016年20期)2016-09-29
快乐作文·低年级(2016年7期)2016-09-22
企业导报(2016年9期)2016-05-26
小小说月刊·下半月(2016年8期)2016-05-14
湖北农业科学(2014年12期)2015-01-06
中学生英语·外语教学与研究(2008年8期)2008-12-19
