
HPLC法测定吡拉西坦颗粒有关物质和含量
2020-11-06 02:01:22聂延君郑静徐玉文
药学研究 2020年10期
聂延君,郑静,徐玉文
(山东省食品药品检验研究院,仿制药研究与评价重点实验室,山东省仿制药一致性评价工程技术研究中心,山东 济南 250101)
吡拉西坦(Piracetam)是一种作用于中枢神经系统的益智药,比利时UCB研究所于1963年合成成功,适用多种原因所致的记忆减退及轻、中度脑功能障碍;也用于儿童智能发育迟缓[1-5]。吡拉西坦颗粒现行标准为国家局WS-10001-(HD)-1132-2002,现行标准中没有有关物质检查,对本品安全性缺少控制。含量测定则采用凯氏定氮法,检验过程中样品处理复杂烦琐,氮消解过程时间长,试验过程中加热温度和时间对反应终点均有较大影响。因此,为提高本品质量,简便操作,本文建立了高效液相色谱(HPLC)法测定吡拉西坦颗粒有关物质和含量,以期为质量标准提高提供技术支持。
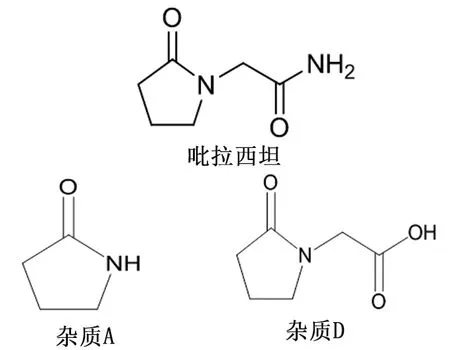
图1 吡拉西坦和相关杂质结构
1 仪器与试药
1.1 仪器 岛津LC-20AT高效液相色谱仪(日本岛津公司),Waters e2695高效液相色谱仪(美国Waters公司)。
1.2 试药 原料药(东北某有限公司,批号:030180672);吡拉西坦颗粒(江西某公司,批号:201752,201822和201824);吡拉西坦对照品(中国食品药品检验研究院,批号:100386-201703,含量:100.0%);杂质A(中国食品药品检验研究院,批号:190154-201501,含量:98.9%);杂质D对照品(EP,批号:2426-054A5,含量:98.1%)。乙腈为色谱纯,盐酸、过氧化氢、磷酸氢二钾均为分析纯。
2 方法与结果
2.1 有关物质检查方法[6]色谱柱为Kromasil 100-5 C18(4.6 nm×250 mm,5 μm);流动相A为0.1%磷酸氢二钾溶液(用磷酸调节pH值至6.0±0.05),流动相B为乙腈,梯度洗脱(0~20 min,95%A→80%A;20~28 min,80%A;28~28.1 min,95%A;28.1~40 min,95%A)。流速为每分钟1.0 mL;检测波长为205 nm,柱温35 ℃。进样量20 μL。系统适用性溶液:取吡拉西坦对照品约25 mg,进行碱破坏后,加杂质A对照品10 μL,稀释至刻度,摇匀。系统适用性要求:杂质D峰相对吡拉西坦峰的保留时间约为0.72,吡拉西坦峰与杂质A峰的分离度应大于3.0,吡拉西坦峰的拖尾因子应小于2.0。供试品溶液浓度为0.5 mg·mL-1,对照溶液浓度为0.5 μg·mL-1。
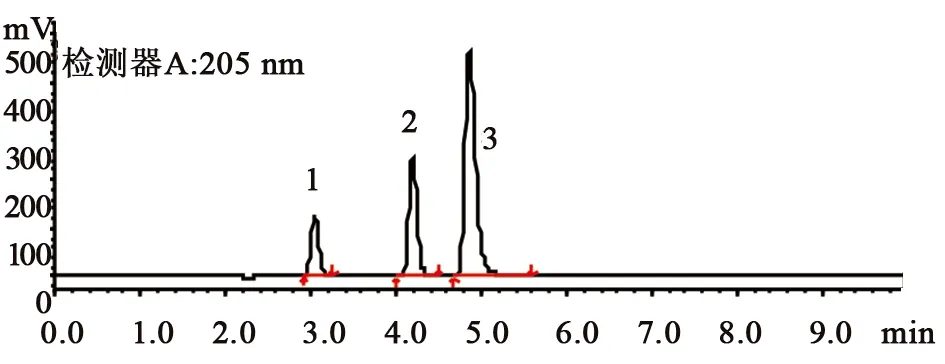
1.杂质D;2.吡拉西坦;3.杂质A图2 系统适用性图谱
2.2 方法学验证
2.2.1 专属性试验 采用强酸、强碱、氧化、加热和光照的方式对吡拉西坦进行破坏性试验,结果各降解杂质与吡拉西坦主峰以及杂质之间的分离度均良好,能够实现有效分离,其中杂质D为主要降解杂质。
2.2.2 精密度试验 取吡拉西坦对照溶液(0.5 μg·mL-1),精密量取20 μL照上述色谱条件连续进样6次,结果峰面积RSD为0.38%,说明该方法的精密度较高。
2.2.3 稳定性试验 取吡拉西坦颗粒供试品溶液(批号:20180202)按“2.1”项下方法制备一份供试品溶液,分别于0、2、4、6、8 h,按上述色谱条件测定,记录峰面积。结果杂质D峰面积RSD为0.18%,其他单杂峰面积RSD为0.05%,总杂质RSD为0.56%,表明供试品溶液在8 h内稳定性良好。
2.2.4 线性关系考察 精密称取吡拉西坦对照品适量制成每1 mL中含吡拉西坦0.10、0.25、0.50、0.75、1.00 μg的溶液,作为线性溶液。照上述色谱条件分别精密量取20 μL进样测定,以浓度为横坐标,峰面积为纵坐标,绘制标准曲线。结果吡拉西坦在0.10~1.00 μg·mL-1范围内线性关系良好(Y=43 829X+1 652.1,相关系数r为0.999 0)。
2.2.5 准确度试验 分别精密称取吡拉西坦对照品适量,按处方量称入空白辅料,加乙腈-水(5∶95)溶解并过滤,取续滤液供试品溶液,分别制成每1 mL含吡拉西坦0.10、0.50、0.75 μg的溶液,每个浓度配制3份,照上述色谱条件精密量取20 μL进样。结果回收率平均值为97.1%。说明该方法的回收率良好。
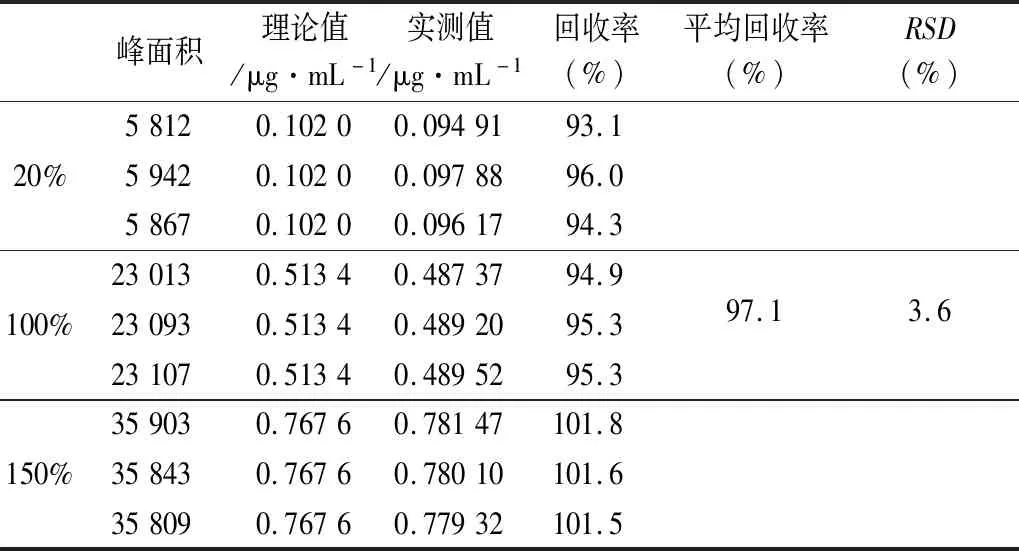
表1 准确度试验结果表
2.2.6 检出限与定量限 分别取吡拉西坦对照品适量,加乙腈-水(5∶95)溶解并逐级稀释,定容,进样,当吡拉西坦峰信噪比(S/N)为3时,即为检出限,结果为0.01 μg·mL-1。在信噪比(S/N)为10时,即为定量限,结果为0.03 μg·mL-1。
2.2.7 耐用性试验 分别采用调整柱温(30、35、40 ℃)、色谱柱和高效液相色谱仪的方法进行耐用性试验,结果,各条件下均能满足系统适用性要求,说明该方法耐用性较好。
2.3 含量测定方法 参照有关物质方法, 采用Kromasil 100-5 C18;流动相为0.1%磷酸氢二钾溶液(用磷酸调节pH值至6.0±0.05)-乙腈(95∶5),由梯度洗脱调整为等度洗脱。检测波长为205 nm,流速为每分钟1.0 mL。供试品和对照品溶液浓度均为0.1 mg·mL-1的溶液。精密量取供试品溶液与对照品溶液各20 μL,分别注入液相色谱仪,记录色谱图,按外标法以峰面积计算。

A.酸破坏;B.碱破坏;C.氧化破坏;D.热破坏;E.光破坏 1.杂质D;2.吡拉西坦;3.其他杂质图3 专属性试验图谱
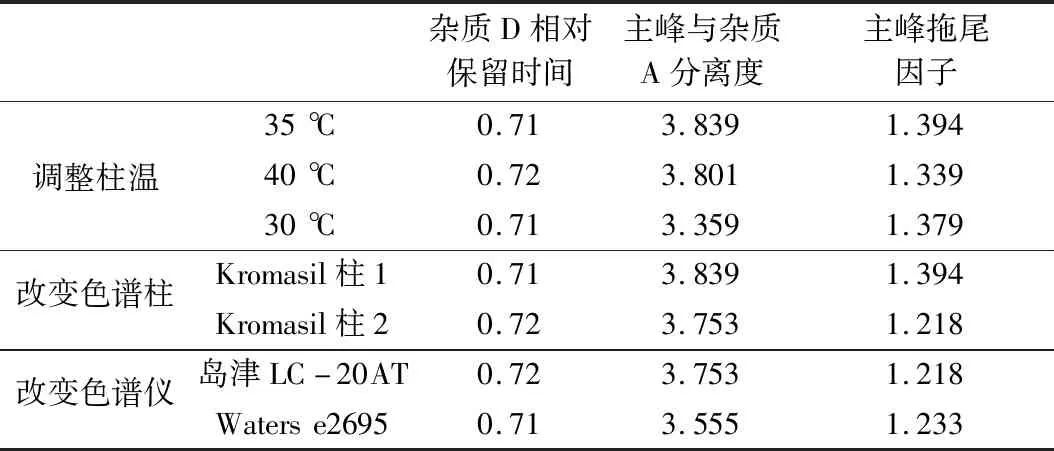
表2 耐用性汇总表
2.4 样品测定 根据“2.1”和“2.3”项下的有关物质和含量测定方法,对样品进行测定,具体结果见表3。
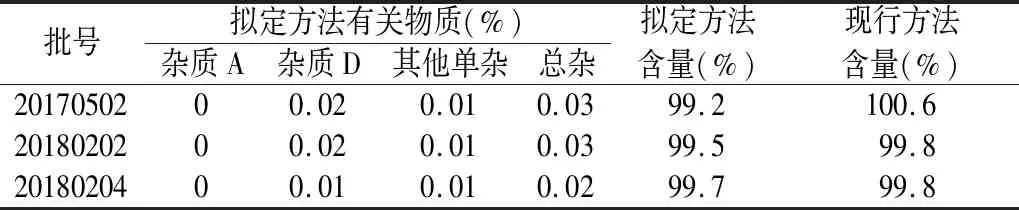
表3 有关物质检验结果汇总表
3 讨论
3.1 吡拉西坦颗粒的每天服用量较大,一般为每天2.4~4.8 g,现行标准没有设定有关物质检查,不能客观反映样品中杂质情况,属于安全性项目缺失,存在用药安全隐患,因此需增订有关物质。对酸、碱、氧化、高温、光照等破坏性试验产生的降解产物分析,杂质D是主要的降解产物,因此杂质D能间接反映样品的生产工艺、处方以及贮存条件变化情况,对吡拉西坦颗粒质量控制具有重要意义,而杂质A为主要的工艺杂质,因此将杂质A和D作为已知杂质进行重点控制。按照ICH杂质限量研究的指导原则,将杂质A、D的限度拟定为不得过0.15%。其他单个杂质不得过0.1%,杂质总和不得过0.5%。
3.2 现行标准中含量测定采用凯氏定氮法,检验过程中样品和空白对照的处理复杂烦琐,消解时间长,试验过程中需控制加热温度和时间,多种因素均可对反应终点有较大影响,容易造成试验结果准确度差的问题。《中国药典》2015年版中吡拉西坦原料及相关制剂(口服溶液、片剂、注射液、胶囊)均采用高效液相色谱法测定含量[7],制剂企业也有升级质量标准的要求。因此,为了统一标准,简便操作,含量测定方法修订为HPLC法,该方法简便、准确、可行,检测结果与现行结果比较,基本一致,因此可用于吡拉西坦颗粒的质量控制。
猜你喜欢
临床儿科杂志(2023年8期)2023-09-29 09:38:52
煤化工(2022年3期)2022-07-08 07:24:42
昆明医科大学学报(2021年10期)2021-12-02 03:24:38
艺术品鉴(2020年6期)2020-12-06 10:49:08
作文周刊·小学四年级版(2018年40期)2018-04-09 08:20:04
领导文萃(2017年6期)2017-03-24 09:31:39
中学生数理化·高一版(2016年7期)2016-12-07 20:47:07
中国资源综合利用(2016年10期)2016-01-22 08:36:09
中学生数理化·中考版(2015年12期)2015-09-10 07:22:44
中国当代医药(2015年30期)2015-03-01 02:08:09
