
IDO抑制剂PCC0208009在结肠癌CT26荷瘤小鼠的PK/PD研究
2020-11-05 12:35:34李晓鹏王洪波田京伟杜广营
烟台大学学报(自然科学与工程版) 2020年4期
姜 雪,李晓鹏,郑 爽,王洪波,田京伟,杜广营
(烟台大学药学院分子药理和药物评价教育部重点实验室,烟台大学新型制剂与生物技术药物研究山东省高校协同创新中心,山东 烟台 264005)
免疫耐受是肿瘤发生与进展的主要原因,也是影响肿瘤临床治疗效果的重要阻碍因素.通过逆转肿瘤免疫耐受,激活机体自身免疫系统清除杀伤肿瘤细胞的免疫治疗已经成为一种成熟的肿瘤治疗手段[1].已有6个免疫检查点类药物被FDA批准上市,用于多种肿瘤如非小细胞肺癌、肾癌、膀胱癌、头颈癌、黑色素瘤等的一线或二线治疗[2].吲哚胺2,3-双加氧酶(Indoleamine 2,3-dioxygenase, IDO, EC 1.13.11.42)是肝脏外唯一催化色氨酸(Tryptophan, Trp)沿犬尿氨酸(Kynurenine, Kyn)途径分解代谢的限速酶[3].近年研究发现IDO与肿瘤免疫耐受密切相关,可通过多种机制介导肿瘤细胞的免疫逃逸,如色氨酸耗竭抑制局部T细胞增殖,代谢产物诱导调节性T细胞增殖,色氨酸代谢产物促进T细胞凋亡等[4-5].IDO在多种肿瘤微环境中高表达,表达水平与患者生存率呈负相关,是显著的不良预后因子[3,6].IDO抑制剂可逆转IDO介导的免疫耐受,激活效应T细胞,改善肿瘤免疫微环境,协同增强其他药物如免疫检查点药物和化疗药物的肿瘤杀伤抑制作用[7].已上市的免疫检查点类药物临床应答率低,只有20%~30%[8],这可能与IDO共表达或诱导IDO表达有关[9].临床试验结果也显示,抑制IDO能有效提升PD-1等免疫检查点药物的疗效[10].IDO是抗肿瘤免疫治疗领域极具潜力的新靶点[3,6],开发IDO抑制剂是免疫检查点药物临床应用的重要补充[11].
PCC0208009是Bristol-Myers Squibb(BMS)公司专利WO2015/031295 A1中的示例1化合物,化学结构如图1所示[12].本实验室前期研究结果表明,PCC0208009是一个高效的IDO小分子抑制剂,可显著增强替莫唑胺在脑胶质瘤荷瘤动物模型的抗肿瘤效应[13].文献资料显示,血清中Kyn和Trp的含量比值Kyn/Trp,可以作为IDO抑制剂临床前药效作用评价的的生物标记物[14-15].因此,本试验在前期研究的基础上,采用结肠癌CT26荷瘤小鼠模型,以Kyn/Trp变化为药效学评价指标,考察PCC0208009在荷瘤小鼠的药代动力学(pharmacokinetics, PK)和药效动力学(Pharmacodynamics, PD)特点及二者的相关性,为后续IDO抑制剂的开发和临床治疗效果的预测提供参考依据.
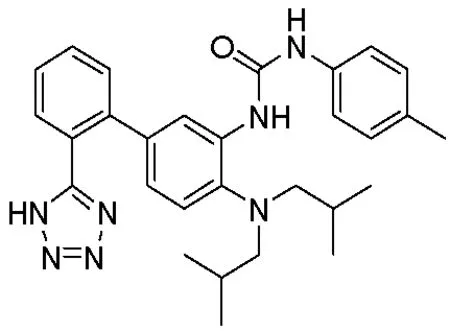
图1 PCC0208009的化学结构式[12]
1 实验材料
1.1 细胞株与实验动物
小鼠结肠癌细胞CT26.WT,由中国科学院典型培养物保藏委员会细胞库提供,使用含10%胎牛血清、100 U/mL青霉素和10 μg/mL链霉素的RPMI-1640完全培养基于37 ℃,5% CO2的环境中培养,隔天进行细胞换液,每2到3 d进行细胞传代,传代比例为1∶2~1∶5.
实验用BALB/c小鼠,雄性,体重21~23 g,购自北京华阜康生物科技股份有限公司,实验动物生产许可证号:SCXK(京)2014-0004.实验动物饲养于SPF级(无特定病原体)环境中,温度为20±2 ℃,相对湿度为30%~70%,人工光照,昼夜12 h交替.所有实验动物相关的实验方案与操作均经过烟台大学实验动物伦理委员会审核及批准.
1.2 供试品与主要试剂
PCC0208009,白色粉末,难溶于水,购自上海瀚香生物科技有限公司,HPLC鉴定纯度大于99.5%,分子式为C29H35N7O,相对分子质量487.63.PCC0208009溶液配制:先加入适量预先配置的溶媒20% Kolliphor®HS 15(Solutol),50 ℃超声至溶液澄清,再用20% Solutol定容至所需体积,配制成不同浓度的药物.Matrigel®基质胶购自美国BD Biosciences公司.
2 实验方法
2.1 CT26荷瘤小鼠制备
收集对数生长期的CT26结肠癌细胞,重悬于无血清培养基中,调整细胞浓度为1×106个/mL,制备好的细胞悬液与Matrigel基质胶等体积混合,细胞终浓度为5×105个/mL.超净工作台中,于每只小鼠背部肩胛处皮下接种0.2 mL肿瘤细胞悬液,接种量为1×105个/只.待瘤块长至500~1000 mm3时,剥出瘤块,无菌手术剪刀剪碎,用皮埋植入穿刺针(直径1.2 mm)再次接种至小鼠背部肩胛处皮下,制备CT26小鼠移植瘤模型.游标卡尺测量肿瘤最大直径(a)和最小直径(b),根据公式V=0.5×a×b2计算肿瘤体积,当肿瘤体积大于300 mm3时,动物分组,用于试验.
2.2 剂量设计
本实验室前期CT26荷瘤小鼠模型药效研究显示,PCC0208009在25~200 mg/kg范围内可剂量依赖性抑制移植瘤的生长,200 mg/kg剂量下,肿瘤生长抑制率已基本达到最大药效水平.因此,在时间效应关系的PK/PD研究中,PCC0208009的剂量设为200 mg/kg,单次灌胃给药后在不同时间点检测肿瘤和血浆中PCC0208009,Kyn和Trp的含量.在剂量效应关系的PK/PD研究中,PCC0208009的剂量设为25、50、100、200、400 mg/kg,单次或多次灌胃给药后4 h,检测肿瘤和血浆中PCC0208009、Kyn和Trp的含量.
2.3 时间效应关系PK/PD研究
荷瘤小鼠根据肿瘤体积随机分成给药前0 h对照组及给药后不同时间点组,每组5只动物.对照组直接采血及瘤块,给药后5 min(0.083 h)、15 min(0.25 h)、30 min(0.5 h)、1 h、2 h、4 h、8 h、12 h、24 h组,分别单次灌胃给予200 mg/kg的PCC0208009,给药体积0.1 mL/10 g,并按时间点采血及瘤块.通过眼眶采血0.5~0.8 mL,肝素抗凝.取血后,小鼠以脱颈椎实施安乐死,手术剥取完整瘤块.
所得抗凝血在4000 r/min离心条件下,离心10 min,并分离得到血浆.采用LC-MS/MS方法,检测瘤块和血浆中PCC0208009,Kyn和Trp含量,并计算主要药代参数和Kyn/Trp.血浆样品检测中,取50 μL小鼠血浆样品,采用HLB小柱进行固相萃取后用已建立并验证的LC-MS/MS替代分析物(Kyn-d4和Trp-d5)法进行Kyn和Trp浓度测定;肿瘤样品检测中,肿瘤组织用1∶4体积的纯净水分散后进行高速匀浆约30 s,取50 μL组织匀浆液,采用HLB小柱进行固相萃取后用已建立并验证的LC-MS/MS替代分析物(Kyn-d4和Trp-d5)法进行Kyn和Trp浓度测定[16-17].
2.4 剂量效应关系PK/PD研究
剂量效应关系PK/PD研究可分为2个部分:单次给药试验和多次给药试验.单次给药或多次给药实验中,荷瘤小鼠均根据肿瘤体积,随机分为6组:对照组,25、50、100、200、400 mg/kg PCC0208009给药组,每组各5只动物.单次给药试验中,小鼠分别单次灌胃给予20% Solutol和相应不同剂量的PCC0208009,给药体积0.1 mL/10 g,给药后4 h,采集血浆和肿瘤组织,LC-MS/MS法检测PCC0208009,Kyn和Trp含量,并计算Kyn/Trp.多次给药试验中,小鼠分别灌胃给予20% Solutol和相应不同剂量的PCC0208009,每天2次,连续3 d,给药体积0.1 mL/10 g,给药后4 h,采集血浆和肿瘤组织,LC-MS/MS法检测PCC0208009,Kyn和Trp含量,并计算Kyn/Trp.
2.5 统计学方法
3 实验结果
3.1 时间效应关系PK/PD研究
单次灌胃给予200 mg/kg的PCC0208009,检测给药后不同时间点血浆和肿瘤中PCC0208009的浓度,计算主要药代参数.药时曲线图见图2(a).血浆和肿瘤中Tmax分别为0.25 h和0.5 h,达峰浓度Cmax分别为13.3 μg/mL和3.86 μg/g,PCC0208009灌胃给药后入血较快,在0.25 h时浓度为13.3 μg/mL,随后血浆中药物浓度快速下降,至24 h时低于检测下限;肿瘤中药物浓度缓慢下降,至24 h时仍可检测到药物.血浆和肿瘤中药时曲线下面积AUC0-t分别为22.9 μg·h/mL和17.5 h·μg/g,半衰期T1/2分别为6.22 h和7.08 h,血浆中药物平均滞留时间MRT0-t为2.84 h.
单次灌胃给予200 mg/kg的PCC0208009, 血浆和肿瘤中的Kyn/Trp变化见图2(b).血浆中,Kyn/Trp在给药后2 h开始显著低于给药前,这种抑制作用可持续至给药后24 h(各时间点P<0.05);肿瘤中,给药后2~8 h,Kyn/Trp显著低于给药前(P<0.05).血浆和肿瘤中Kyn/Trp的最大抑制率分别为46.7%和62.1%,均出现在给药后8 h.单次灌胃给药200 mg/kg后2~8 h内,PCC0208009可同时显著降低Kyn/Trp,有效抑制血浆和肿瘤中IDO的活性.

和0 h对照组数据比较.
3.2 单次给药剂量效应关系PK/PD研究
根据时间效应关系研究结果,荷瘤小鼠单次灌胃给予25~400 mg/kg PCC0208009,在给药后4 h,检测血浆和肿瘤中药物浓度以及Kyn/Trp比值的变化.血浆和肿瘤中药物浓度见图3(a),在25~400 mg/kg剂量范围内,血浆和肿瘤中药物浓度均随剂量的增大而增加,呈剂量依赖性.血浆和肿瘤中Kyn/Trp改变见图3(b),血浆中,PCC0208009可剂量依赖性降低Kyn/Trp,各个剂量水平Kyn/Trp均显著低于对照组(P<0.05);肿瘤中,PCC0208009也可剂量依赖性的降低Kyn/Trp,但25 mg/kg剂量下Kyn/Trp与对照组相比无统计学差异(P>0.05),50~400 mg/kg各个剂量水平Kyn/Trp均显著低于对照组(P<0.05).血浆和肿瘤中,给药剂量200 mg/kg时,实现对Kyn/Trp的半数抑制,给药剂量为400 mg/kg时,对Kyn/Trp的抑制率无明显升高,给药剂量为200 mg/kg和400 mg/kg时,抑制率分别为47.0% vs 49.3% 和52.2% vs 51.0%.
3.3 多次给药剂量效应关系PK/PD研究
荷瘤小鼠在多次灌胃给予25~400 mg/kg PCC0208009,给药后4 h,检测药物浓度和Kyn/Trp比值变化.血浆和肿瘤中药物浓度见图4(a).在25 ~400 mg/kg剂量范围内,血浆和肿瘤中药物浓度均随剂量的增大而增加,呈剂量依赖性.血浆和肿瘤中Kyn/Trp改变见图4(b).血浆中,PCC0208009可剂量依赖性降低Kyn/Trp,各个剂量水平Kyn/Trp均显著低于对照组(P<0.05);肿瘤中,PCC0208009也可剂量依赖性的降低Kyn/Trp,各个剂量水平Kyn/Trp均显著低于对照组(P<0.05).血浆和肿瘤中,200 mg/kg和400 mg/kg 剂量组的Kyn/Trp降低相当,抑制率分别为57.7% vs 61.5% 和72.0% vs 73.6%.

和对照组数据比较.

和对照组数据比较.
4 讨 论
本实验室前期对PCC0208009进行了部分体内外活性研究,表明其是一个高效的IDO小分子抑制剂,可显著增强替莫唑胺在脑胶质瘤荷瘤动物模型的抗肿瘤效应[13].本试验是在前期研究的基础上,进一步对其在荷瘤小鼠体内的PK/PD行为特点进行探讨.IDO抑制剂属于肿瘤免疫增强药物,大量文献资料显示其在荷瘤小鼠体内的抗肿瘤作用并不强,肿瘤抑制率约30%~50%[18],即采用肿瘤抑制率作为药效指数对IDO抑制剂来说不敏感.前期研究和文献报道提示,血清中Kyn和Trp的含量的比值Kyn/Trp可以作为评价IDO抑制剂的药效作用的生物标记物.因此,本研究中采用Kyn/Trp的改变作为药效指标进行PK/PD关系研究.
时间效应关系PK/PD研究结果表明,单次灌胃给药后,PCC0208009快速在血浆中分布,Tmax为0.25 h,肿瘤组织中分布稍慢,Tmax为0.5 h,达峰后血浆中药物浓度快速下降,而肿瘤中药物浓度下降缓慢.给药后2~8 h内,PCC0208009可显著降低血浆和肿瘤内的Kyn/Trp,抑制IDO的活性,血浆和肿瘤中Kyn/Trp的最大抑制率均出现在给药后8 h.对比血浆和肿瘤中PCC0208009药物浓度和Kyn/Trp抑制率,药物发挥最大抑制活性时,药物浓度并非最高,可知PCC0208009对IDO的抑制作用具有一定的滞后性,分析可能与采用的药效指标Kyn和Trp是IDO酶活的间接作用结果有关.
根据时间效应关系研究结果,单次和多次给药试验中,在给药4 h时,观察不同药物剂量下,PCC0208009的PK/PD相关性.结果均显示,在25 ~400 mg/kg剂量范围内,血浆和肿瘤中药物浓度均随剂量的增大而增加,呈剂量依赖性.PCC0208009也可剂量依赖性降低血浆和肿瘤中Kyn/Trp.多次给药试验中,25 mg/kg剂量下,血浆和肿瘤中Kyn/Trp均显著低于对照组,可以认为25 mg/kg是PCC0208009在该模型的药效起始剂量.无论单次给药还是多次给药,血浆和肿瘤中,200 mg/kg和400 mg/kg 剂量组的Kyn/Trp降低作用相当,即200 mg/kg 剂量下,PCC0208009已经达到最大药效水平,与前期荷瘤小鼠体内药效研究试验结果一致,为后续IDO抑制剂的开发提供了参考依据.
5 结 论
IDO抑制剂PCC0208009在结肠癌CT26荷瘤小鼠体内的PK/PD研究表明,在给药后2~8 h内PCC0208009可有效抑制血浆和肿瘤中Kyn/Trp,药物在血浆和肿瘤中的浓度及对Kyn/Trp的抑制作用均呈剂量依赖性,PK和PD具有较好的相关性.本试验条件下,PCC0208009的药效起效剂量为25 mg/kg,200 mg/kg时已接近最大药效平台.本研究可为IDO抑制剂的开发以及预测IDO抑制剂的临床治疗效果提供理论基础.
猜你喜欢
天然产物研究与开发(2018年8期)2018-09-10 05:48:18
中成药(2018年3期)2018-05-07 13:34:35
上海农业学报(2017年3期)2017-04-10 12:39:26
中国比较医学杂志(2017年3期)2017-01-17 05:40:54
中国中医药信息杂志(2016年7期)2016-12-01 06:07:46
天然产物研究与开发(2016年6期)2016-06-05 10:29:30
山东医药(2015年15期)2016-01-12 00:39:58
中国当代医药(2015年16期)2015-03-01 02:03:13
中国药理学通报(2014年2期)2014-05-09 08:22:39
山东畜牧兽医(2014年10期)2014-04-05 14:53:34
