
藤茶高通量转录组分析及黄酮类化合物合成相关基因挖掘
2020-11-02 02:41许明杨志坚黄学敏郑金贵
南方农业学报 2020年8期
许明 杨志坚 黄学敏 郑金贵

摘要:【目的】分析藤茶高通量轉录组序列,从中挖掘出黄酮类化合物合成相关基因,为进一步揭示藤茶黄酮类化合物生物合成调控机制提供理论参考。【方法】分别采集藤茶的幼叶和成熟叶,提取其总RNA构建cDNA文库,采用Illumina HiSeqTM 4000高通量测序平台对藤茶叶片进行转录组测序,经过滤处理后运用Trinity组装,将获得的Unigene与Nr、Nt、Pfam、Swiss-Prot、GO、KO和KOG 7个数据库进行比对注释,并预测Unigenes的编码区序列(CDS);基于KEGG信号通路富集分析,发掘藤茶黄酮类化合物合成相关基因。【结果】藤茶叶片转录组测序获得82126236条原始测序序列(Raw reads),过滤处理后得到80156972条高质量序列(Clean reads),进一步组装拼接得到92472条Unigenes,平均长度为1208 bp,N50长度为1780 bp,其中,至少在1个数据库注释的Unigenes有84217条,占Unigenes总数的91.07%,有8944条Unigenes在7个数据库均被注释,占Unigenes总数的9.67%。在GO数据库成功注释的41116条Unigenes可分为生物学过程、细胞组分和分子功能三大类,共56个小类;在KOG数据库注释的14553条Unigenes可分成25类,其中,一般功能预测注释成功的Unigenes最多(1946条);其次是翻译后修饰、蛋白质翻转、分子伴侣(1776条),参与次生代谢物质的生物合成、转运和降解的Unigenes较少,仅有319条;KEGG信号通路富集分析发现,共有15262条Unigenes注释到128条KEGG信号通路,以注释为代谢的Unigenes最多,为8694条,其中筛选获得有98个黄酮类化合物合成相关基因,分别编码苯丙烷代谢通路的3种关键酶和类黄酮代谢通路的14种关键酶。藤茶叶片转录组Unigenes与Swiss-Prot和Nr数据库比对,获得52582条CDS序列,ESTScan 3.0.3预测获得35535条CDS序列。【结论】藤茶在细胞过程、代谢过程、单有机体过程、细胞和细胞部分、结合和催化活性能力分布的基因较丰富,在一般功能、翻译、翻译后修饰、蛋白质翻转及分子伴侣的基因表达量较高,具有较强的碳水化合物代谢能力。多种关键酶基因参与藤茶黄酮类化合物的生物合成,推测其生物合成途径存在多条分支,调控机制也较复杂。
关键词: 藤茶;转录组;黄酮类化合物;基因挖掘;高通量测序
中图分类号: S571.1 文献标志码: A 文章编号:2095-1191(2020)08-1797-09
Transcriptome analysis of Ampelopsis grossedentata (Hand. Mazz.) W. T. Wang and mining of putative genes involved in flavonoid biosynthesis
XU Ming1,2, YANG Zhi-jian1,2, HUANG Xue-min3, ZHENG Jin-gui1,2*
(1Key Laboratory of Crop Biotechnology, Fujian Province University, Fuzhou 350002, China; 2College of Agriculture,Fujian Agriculture and Forestry University, Fuzhou 350002, China; 3Forestry Bureau of Yongchun County,
Fujian Province, Yongchun, Fujian 362600)
Abstract:【Objective】The study aimed to obtain the high throughput transcriptome sequence information and explore the genes related to the flavonoid biosynthesis in Ampelopsis grossedentata(Hand. Mazz.) W. T. Wang. It provided the theoretical basis for further revealing the biosynthetic pathway and regulation mechanism of flavonoids in A. grosse-dentata. 【Method】The young and mature leaves of A. grossedentata were collected respectively for the extraction of total RNA which was used to construct cDNA library. The transcriptome of A. grossedentata leaves was sequenced using an Illumina HiSeqTM 4000 high throughput platform. The sequences were assembled with Trinity after data filtering. The obtained Unigenes were compared and annotated with seven databases, including Nr, Nt, Pfam, Swiss-Prot, GO,KO and KOG, and the coding region sequences(CDS) were predicted by Blast and ESTScan software. KEGG signal pathway enrichment analysis was used to identify genes related to flavonoid synthesis in A. grossedentata. 【Result】A total of 82126236 raw reads were obtained with the transcriptome sequencing. After reads filtering, a total of 80156972 clean reads were obtained, which were used to assemble and merge into 92472 Unigenes with an average length of 1208 bp and a N50 length of 1780 bp.Of these unigenes, 84217(91.07%) were successfully annotated in at least one of the Nr, Nt, Swiss-Prot, KEGG, COG and GO databases, and 8944 unigenes(9.67%) were annotated in all databases. Among them,41116 Unigenes annotated in GO database were matched to 56 sub-classes of biological function, cell component and molecular function,and 14553 unigenes annotated in KOG database were divided into 25 sub-classes. The number of the unigenes annotated in the general function was the greatest(1946); the post translational modification, protein turn over and chaperones was the second(1776); the unigenes involved in the biosynthesis of secondary metabolites, transportation and degradation were less(only 319). The KEGG signal pathway enrichment analysis results showed that a total of 15262 unigenes were participated in 128 KEGG pathways. The unigenes annotated with metabolism were the greatest in number(8694), 98 of which participated in the flavonoid synthesis pathway of A. grossedentata. They encoded three key enzymes of phenylpropane metabolic pathway and 14 key enzymes of flavonoid metabolic pathway. By comparing with Swiss-prot and Nr database, 52582 CDS were obtained. A total of 35535 CDS sequences were obtained via ESTScan 3.0.3 prediction. 【Conclusion】A. grossedentata has abundant genes related to cell process, metabolic process, single organism process, cell and cell part, binding and catalytic activities. It shows a high level of gene expression in general function, translation, post translational modification, protein turn over, chaperones.And it has a strong carbohydrate metabolic ability as well. Many key enzymes are involved in the biosynthesis of flavonoids in A. grossedentata, suggesting that there might be multiple branches in the biosynthetic pathway, and the regulation mechanisms might also be complex.
Key words: Ampelopsis grossedentata(Hand. Mazz.) W. T. Wang; transcriptome; flavonoids; gene mining; high-throughput sequencing
Foundation item: National Science and Technology Support Program(2013BAD01B05);Science and Technology Innovation Project of Fujian Agriculture and Forestry University(KFA17424A)
0 引言
【研究意义】藤茶[Ampelopsis grossedentata(Hand.Mazz.) W. T. Wang]是我国栽培历史悠久的一种茶药两用植物,其主要药效成分为黄酮类化合物,具有良好的开发利用前景(何桂霞等,2007;冉京燕等,2016)。但由于藤茶基因组及转录组数据缺乏,遗传背景尚不清楚,严重制约了藤茶种质资源鉴定和保护、遗传多样性分析及分子育种等工作的开展。此外,产地、栽培条件等诸多因素均会导致藤茶黄酮含量存在明显差异,从而影响藤茶产品的质量和疗效(王家胜等,2014;冉京燕等,2016)。因此,开展藤茶转录组及其黄酮类化合物的代谢途径研究,了解藤茶遗传信息及其药效成分合成的分子调控机制,对藤茶种质资源的持续高效利用及药用品质提升具有重要意义。【前人研究进展】迄今,国内外针对藤茶的研究主要集中在化学成分的分离鉴定及药理作用等方面(Zhang et al.,2006;谭沙等,2015),其基因水平的相关研究仅有少量报道。黄海波(2008)从生理生化层面分析了藤茶不同季节苯丙氨酸解氨酶(PAL)活性与藤茶黄酮含量变化的相关性;付明等(2013)利用RT-PCR克隆藤茶查尔酮合成酶(CHS)基因;许明等(2017)筛选了适用于藤茶不同组织实时荧光定量PCR分析的内参基因。转录组测序是指利用第二代高通量测序技术进行cDNA测序,能在无参考基因组信息的情况下,对某一物种的转录组进行全面分析,现已成为揭示特定生物学过程分子机制的一种有效手段(Wilhelm and Landry,2009;严志祥等,2019)。目前,利用高通量测序技术已完成较多药用植物的转录组分析,并鉴定出大量与黄酮类化合物合成相关的基因。马婧等(2016)对草麻黄进行转录组测序分析,鉴定出黄酮类化合物代谢相关基因70个;刘伟等(2018)对银杏不同生长时期叶片进行转录组测序分析,筛选出50个银杏类黄酮生物合成相关的候选基因;邹福贤等(2019)对3个生长时期的金线莲进行转录组测序,找出22个黄酮类代谢通路上的差异表达基因,并结合其黄酮类成分,绘制出相应的生物合成途径;Yang等(2019)对不同季节的大叶蛇葡萄叶片进行转录组分析,鉴定出31个类黄酮生物合成相关基因和50个黄酮转运和生物修饰相关基因。【本研究切入点】至今,鲜见有关藤茶转录组测序及其黄酮类化合物合成相关基因挖掘的研究报道。【拟解决的关键问题】利用Illumina HiseqTM 4000高通量测序平台对藤茶叶片进行转录组测序,将获得的序列信息进行拼接和组装,并利用生物大分子公共数据库对所得基因进行功能注释和分类,挖掘出藤茶黄酮类化合物生物合成相关基因,为进一步揭示黄酮类化合物生物合成调控机制提供理论参考。
1 材料与方法
1. 1 试验材料
供试藤茶引自福建尤溪县下村林场。Qubit RNA Assay Kit购自美国Invitrogen公司。主要仪器设备:NanoDrop ND2000超微量分光光度计(Thermo,美国)、Agilent 2100生物分析仪(Agilent,美国)。
1. 2 总RNA提取
分别采集藤茶的幼叶和成熟叶,参照许明等(2015)的方法提取其总RNA,并采用1.0%琼脂糖凝胶电泳进行检测。利用Qubit RNA Assay Kit测定RNA浓度。
1. 3 cDNA文库构建及测序
将上述2种叶片RNA等比例混合成1 mg样品,以干冰保存送至北京诺禾致源生物信息科技有限公司进行cDNA文库构建,并采用Illumina HiSeqTM 4000高通量测序平台进行转录组测序。
1. 4 转录组组装
对测序获得的原始测序序列(Raw reads)进行过滤处理,得到高质量序列(Clean reads),然后采用Trinity对其进行拼接,将得到的转录本(Transcript)作为参考序列,并取每条基因中最长的转录本作为Unigene,用于后续转录组序列分析。
1. 5 基因功能注释和分類
为获得全面的基因功能信息,将拼接得到的Unigene分别在Nr、Nt、Pfam、Swiss-Prot、GO、KO和KOG等7个数据库中进行功能注释和分类,以及进行KEGG信息通路富集分析。
1. 6 编码区序列(CDS)预测及分析
按照Swiss-Prot和Nr数据库的优先级顺序,将藤茶叶片转录组Unigene进行BLAST比对。从比对上的序列中提取出Transcript开放阅读框(ORF),并将CDS序列翻译成氨基酸序列。未比对上或比对上未预测出结果的序列则采用ESTScan 3.0.3预测其ORF,根据CDS序列翻译获得氨基酸序列。
2 结果与分析
2. 1 转录组测序及组装结果
藤茶叶片转录组测序获得82126236条Raw reads,过滤处理后得到80156972条Clean reads,有效测序长度达12.02 Gb,碱基错误率为0.02%,Q20和Q30分别为97.30%和92.97%,GC含量为43.36%,说明测序质量好,可用于后续分析。
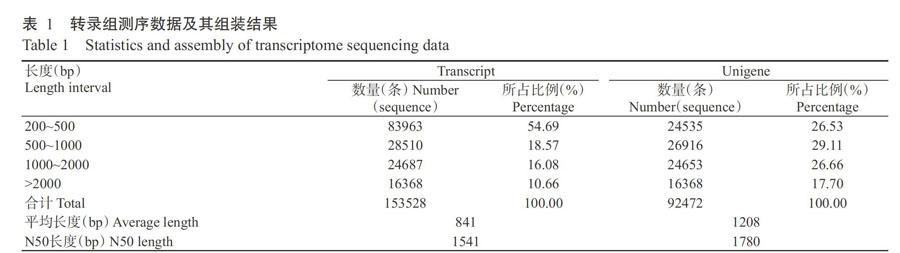
采用Trinity将所有Clean reads拼接得到153528条转录本,平均长度为841 bp,N50为1541 bp,其中,以长度200~500 bp的序列数量最多,为83963条,占54.69%;以长度>2000 bp的序列数量最少,为16368条,占10.66%。将转录本进一步组装拼接得到92472条Unigenes,平均长度为1208 bp,N50为1780 bp,其中,以长度500~1000 bp的序列数量最多,为26916条,占29.11%;以长度>2000 bp的序列数量最少,为16368条,占17.70%(表1)。
2. 2 Unigene功能注释结果
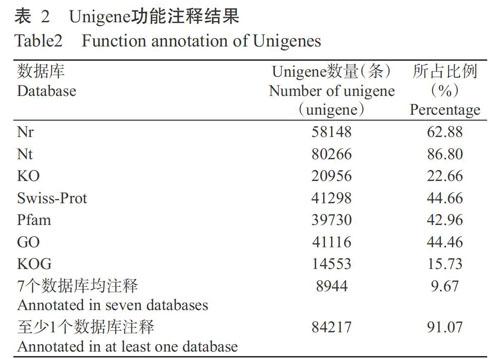
将92472条Unigenes分别在7个数据库进行比对注释,每个数据库中注释成功的Unigenes数量及其占Unigenes总数的比例如表2所示。在7个数据库中,至少在1个数据库注释的Unigenes有84217条,占Unigenes总数的91.07%,其中,在Nt数据库中注释的Unigenes最多(80266条),占总数的86.80%,其次是Nr数据库(58148条),占Unigenes总数的62.88%,KOG数据库中注释的Unigenes数量最少(14553条),占Unigenes总数的15.73%;有8944条Unigenes在7个数据库均被注释,占Unigenes总数的9.67%。
基于Nr数据库比对注释的结果,统计并绘制比对上的物种分布情况(图1),发现注释到葡萄的Unigenes数量最多,所占比例高达88.17%,远高于可可(1.17%)、莲(1.11%)、麻风树(0.69%)、甜橙(0.60%)及其他物种(8.27%),究其原因可能是藤茶与葡萄同属于葡萄科植物,亲缘关系更近。
2. 3 Unigene的GO功能分类结果
从图2可知,在GO数据库成功注释的41116条Unigenes可分为生物学过程、细胞组分和分子功能三大类,共56个小类。由于存在同一个Unigene注释到不同功能的情况,有106044条Unigenes注释归属于生物学过程,其中,以细胞过程、代谢过程和单有机体过程最多;有66390条Unigenes注释归属于细胞组分,其中以细胞和细胞部分最多;有51272条Unigenes注释归属于分子功能,其中,主要以结合和催化活性为主。
2. 4 Unigene的KOG功能分类结果
通过KOG注释可将14553条Unigenes分成25类(图3),其中,一般功能预测注释成功的Unigenes最多(1946条);其次是翻译后修饰、蛋白质翻转、分子伴侣(1776条);翻译、核糖体结构和生物起源注釋成功的Unigenes有1250条;参与次生代谢物质的生物合成、转运和降解的Unigenes较少,仅有319条。此外,有988条未知功能的Unigenes有待进一步研究。
2. 5 Unigene的KEGG信号通路富集分析结果
利用KEGG数据库中的通路数据库(Pathway databases)对Unigene的功能进行富集分析(表3),结果发现共有15262条Unigenes能注释到128条KEGG信号通路。这些通路可分成细胞进程、环境信号处理、遗传信息处理、代谢和有机系统五大类。其中,注释为代谢的Unigenes最多,为8694条,占注释Unigenes总数的56.97%,其次是注释为遗传信息处理的Unigenes,为4120条,占注释Unigenes总数的26.99%。
KEGG信号通路富集分析结果(表3)表明,在五大类通路中,与代谢有关的通路最多,有98条,占所有通路的76.56%,其中,Unigene注释最多为碳水化合物(1671条),其次是氨基酸代谢(1176条)。有512条Unigenes参与苯丙素类、生物碱、黄酮、花青素等生物合成相关的11个其他次生代谢标准通路。
2. 6 Unigene的CDS序列预测结果
按照Swiss-Prot和Nr数据库的优先级顺序,将藤茶叶片转录组Unigene在这2个蛋白数据库进行BLAST比对,获得52582条CDS序列;将未比对上或比对上未预测出结果的序列采用ESTScan 3.0.3进行CDS预测,结果获得35535条CDS序列。在获得的88117条CDS序列中,以长度0~200 nt的CDS序列最多,有52828条;其次是长度201~400和401~600 nt的CDS序列,分别为20564和8796条,其余长度范围的CDS序列均不足4000条(图4)。
2. 7 黄酮类物质生物合成相关基因挖掘及分析结果
黄酮类化合物的主要合成途径:首先通过苯丙烷途径将苯丙氨酸转化为香豆酰-CoA,然后香豆酰-CoA进入黄酮合成途径,与3分子丙二酰CoA结合生成查尔酮,再经分子内的环化反应生成二氢黄酮类化合物,最后通过分支途径合成黄酮、异黄酮、黄酮醇、黄烷醇和花色素等黄酮类化合物(邹丽秋等,2016)。基于KEGG信号通路富集分析结果,共筛选获得98个藤茶黄酮类化合物合成相关基因(表4),其中18个映射到苯丙烷代谢通路上,分别编码苯丙氨酸向香豆酰-CoA转化所需的3种关键酶:苯丙氨酸解氨酶(PAL)、肉桂酸-4-羟化酶(C4H)和对香豆酸—辅酶A连接酶(4CL);有80个Unigenes映射到类黄酮类化合物代谢通路上,编码该通路中的14种关键酶,包括从香豆酰-CoA向二氢黄酮转化所需的查尔酮合成酶(CHS)和查耳酮异构酶(CHI),以及从二氢黄酮到黄酮醇和黄烷醇合成支路所需的黄烷酮-3-羟化酶(F3H)、类黄酮-3-羟化酶(F3H)、类黄酮-3,5-羟化酶(F35H)、黄酮醇合成酶(FLS)、二氢黄酮醇还原酶(DFR)、花青素还原酶(ANR)、无色花色素还原酶(LAR)、无色花色素双加氧酶(LDOX)、香豆酰酯3羟化酶(C3H)、咖啡酰辅酶A-O甲基转移酶(CCOAOMT)、黄酮醇3-甲基转移酶(F3OMT)和莽草酸羟基肉桂转移酶(HCT)共12种。
3 讨论
本研究利用第二代高通量测序技术对藤茶叶片转录组进行测序分析,共获得92472条Unigenes。与其他药用植物比较,藤茶转录组测序得到的Unigenes多于银杏(刘伟等,2018)、大叶蛇葡萄(Yang et al.,2019)和金线莲(邹福贤等,2019)等,与山茱萸(朱畇昊等,2017)和掌叶大黄(李欢等,2018)相当,究其原因可能与物种基因型和取材等因素有关。从Unigene长度来看,本研究得到Unigene的平均长度为1208 bp,N50為1780 bp,二者均明显高于连翘(861 bp,1497 bp)(王兴春等,2015)、草麻黄(453 bp,525 bp)(马婧等,2016)及短丝木犀(697 bp,1200 bp)(陈林等,2016)等,与樱椒树(1116 bp,1768 bp)(Zhou et al.,2015)和掌叶大黄(1108 bp,1794 bp)(李欢等,2018)等相近。N50是评价组装序列完整性的重要指标,其数值越大表示组装得到的长片段越多,当N50≥800 bp说明组装得到序列完整性较好(胡俊杰等,2017)。本研究中N50>800 bp,表明藤茶转录组组装后得到的序列完整性较好,能满足转录组分析的要求,从而保证功能注释能得到较理想的比对结果。
本研究将获得的92472条Unigenes在7个Nr数据库中进行比对注释,结果显示,有84217条Unigenes(91.07%)至少在1个数据库被注释,有8944条Unigenes(9.67%)在7个数据库均被注释,且有20956条(22.66%)、41116条(44.46%)和14553条(15.73%)分别在KO、GO和KOG数据库被注释。从本研究Nr数据库同源比对匹配的物种分布结果可知,注释到葡萄的Unigenes最多,所占比例高达88.17%。与其他药用植物相比,藤茶转录组Unigenes的注释率(91.07%)明显高于草麻黄(81.81%)(马婧等,2016)、银杏(81.67%)(刘伟等,2018)和夏枯草(53.13%)(朱畇昊等,2019)等,可能是因为藤茶为葡萄的近缘物种,在现有数据库葡萄的蛋白序列信息较丰富,导致藤茶Unigenes注释率也较高。
黄酮类化合物的种类较多、分布较广,其生物合成途径目前已基本清楚,各步骤催化反应的所需酶及其基因均有大量研究(Hichri et al.,2011;邹丽秋等,2016)。藤茶是富含黄酮类化合物的一种天然植物,其嫩茎叶中的总黄酮含量高达43.4%~45.52%,远高于其他植物,且藤茶中的黄酮类化合物种类多样,除二氢杨梅素外,还含有其他多种黄酮类成分,如槲皮素、花旗松素、山奈酚、藤茶素、杨梅苷、橙皮素、洋芹素、大黄素和阿福豆素等(Zhang and Yang,2001;何桂霞等,2007;付明等,2015)。本研究对藤茶转录组Unigenes进行KEGG信号通路富集分析,结果发现在五大类通路中,有98条与代谢有关的通路,占所有通路的76.56%,通过进一步筛选,共获得98个黄酮类化合物合成相关的基因,分别编码17种关键酶,包括3个苯丙烷代谢途径的所需酶(PAL、4CL和C4H)和14个类黄酮代谢途径的所需酶(CHS、CHI、F3H、F3H、F35H、FLS、DFR、ANR、LDOX、LAR、F3OMT、HCT、C3H和CCOAOMT),为深入研究藤茶黄酮生物合成途径及其相关关键基因的筛选、功能鉴定及调控机制打下了基础。今后还将通过实时荧光定量PCR等技术检测上述关键酶基因的差异表达情况,同时结合药效成分含量分析,解析上述基因在藤茶药效成分生物合成过程的功能。
4 结论
藤茶在细胞过程、代谢过程、单有机体过程、细胞和细胞部分、结合和催化活性能力分布的基因较丰富,在一般功能、翻译、翻译后修饰、蛋白质翻转及分子伴侣的基因表达量较高,具有较强的碳水化合物代谢能力。多种关键酶基因参与藤茶黄酮类化合物的生物合成,推测其生物合成途径存在多条分支,调控机制也较复杂。
参考文献:
陈林,李龙娜,戴亚平,杨国栋. 2016. 短丝木犀转录组测序及类胡萝卜素生物合成相关基因表达分析[J]. 南京林业大学学报(自然科学版),40(5):21-28. [Chen L,Li L N,Dai Y P,Yang G D. 2016. De novo transcriptome sequencing and analysis of carotenoids biosynthesis related gene expression in Osmanthus serrulatus[J]. Journal of Nanjing Forestry University(Natural Sciences Edition),40(5):21-28.]
付明,黎晓英,王登宇,郭孟齐. 2015. 显齿蛇葡萄叶中黄酮类化合物的研究[J]. 中国药学杂志,50(7):574-578. [Fu M,Li X Y,Wang D Y,Guo M Q. 2015. Flavonoid constituents of leaves of Ampelopsis grossedentata(Hand-Mazz) W. T. wang[J]. Chinese Pharmaceutical Journal,50(7):574-578.]
付明,魏麟,余娟,余小林. 2013. 显齿蛇葡萄查耳酮合成酶基因cDNA克隆及蛋白质序列分析[J]. 中草药,44(1):85-89. [Fu M,Wei L,Yu J,Yu X L. 2013. cDNA cloning and protein sequence analysis of chalcone synthase gene in leaves of Ampelopsis grossedentata[J]. Chinese Traditional and Herbal Drugs,44(1):85-89.]
何桂霞,裴刚杜,方麓,欧阳文,李斌. 2007. 藤茶化学成分的研究[J]. 中国现代中药,9(12):11-13. [He G X,Pei G D,Fang L,Ouyang W,Li B. 2007. Studies on chemical constituents of Ampelopsis grossdentata[J]. Modern Chinese Medicine,9(12):11-13.]
胡俊杰,孟翔,周佳滨,杨陆兴,刘善海,李润钊. 2017. 扶桑綿粉蚧转录组分析[J]. 昆虫学报,60(1):9-17. [Hu J J,Meng X,Zhou J B,Yang L X,Liu S H,Li R Z. 2017. Transcriptome analysis of the cotton mealybug,Phenacoccus solenopsis(Hemiptera:Pseudococcidae)[J]. Acta Entomologica Sinica,60(1):9-17.]
黄海波. 2008. 藤茶PAL特性及其与黄酮和DMY含量变化相关性的研究[D]. 武汉:华中农业大学. [Huang H B. 2008. Studies on the characters of PAL and its correlation between flavones and DMY on contents diversification in Tengcha[D]. Wuhan:Huazhong Agricultural University.]
李欢,张娜,李依民,黑小斌,李元敏,邓翀,颜永刚,刘蒙蒙,张岗. 2018. 利用转录组测序挖掘掌叶大黄蒽醌类生物合成相关基因[J]. 药学学报,53(11):1908-1917. [Li H,Zhang N,Li Y M,Hei X B,Li Y M,Deng C,Yan Y G,Liu M M,Zhang G. 2018. High-throughput transcripto-mic sequencing of Rheum palmatum L. seedlings and elucidation of genes in anthraquinone biosynthesis[J]. Acta Pharmaceutica Sinica,53(11):1908-1917.]
刘伟,王俊燚,李萌,董金金,何崇单,汪贵斌,郁万文,王义强. 2018. 基于转录组测序的银杏类黄酮生物合成关键基因表达分析[J]. 中草药,49(23):5633-5639. [Liu W,Wang J Y,Li M,Dong J J,He C D,Wang G B,Yu W W,Wang Y Q. 2018. Transcriptome sequencing analysis of gene expression of flavonoid biosynthesis in Ginkgo biloba[J]. Chinese Traditional and Herbal Drugs,49(23):5633-5639.]
马婧,成铁龙,孙灿岳,邓楠,史胜青,江泽平. 2016. 草麻黄高通量转录组分析及黄酮类代谢途径相关基因的鉴定[J]. 浙江农业学报,28(4):609-617. [Ma J,Cheng T L,Sun C Y,Deng N,Shi S Q,Jiang Z P. 2016. Characterization of transcriptome reveals pathway of flavonoids in Ephedra sinica Stapf[J]. Acta Agriculturae Zhejiangensis,28(4):609-617.]
冉京燕,方建国,谢雪佳,熊微,王文清. 2016. 藤茶的本草资源学研究概况[J]. 中草药,47(20):3728-3735. [Ran J Y,Fang J G,Xie X J,Xiong W,Wang W Q. 2016. Scientific research on herbal resource of vine tea[J]. Chinese Traditional and Herbal Drugs,47(20):3728-3735.]
谭沙,罗静,李建新,宋珊珊. 2015. 藤茶研究进展[J]. 安徽农业科学,43(31):71-75. [Tan S,Luo J,Li J X,Song S S. 2015. Research progress of vine tea[J]. Journal of Anhui Agricultural Sciences,43(31):71-75.]
王家胜,何磊磊,张妮,孔琪,玉秋萍,余正文. 2014. 不同产地显齿蛇葡萄中二氢杨梅素测定[J]. 中成药,36(1):145-147. [Wang J S,He L L,Zhang N,Kong Q,Yu Q P,Yu Z W. 2014. Dihydromyricetin in Ampelosis grossedentata collected from different habitats[J]. Chinese Traditional Patent Medicine,36(1):145-147.]
王兴春,谭河林,陈钊,孟令芝,王文斌,范圣此. 2015. 基于RNA-Seq技术的连翘转录组组装与分析及SSR分子标记的开发[J]. 中国科学(生命科学),45(3):301-310. [Wang X C,Tan H L,Chen Z,Meng L Z,Wang W B,Fan S C. 2015. Assembly and characterization of the transcriptome and development of SSR markers in Forsythia suspensa based on RNA-Seq technology[J]. Scientia Sinica(Vitae),45(3):301-310.]
许明,杨志坚,郑金贵. 2015. 藤茶叶片总RNA的提取及苯丙氨酸解氨酶基因(pal)片段的克隆[J]. 生物学杂志,32(2):96-99. [Xu M,Yang Z J,Zheng J G. 2015. Extraction of total RNA from leaves of Ampelopsis grossedentata and cloning of phenylalanine ammonia lyase gene fragment[J]. Journal of Biology,32(2):96-99.]
许明,伊恒杰,赵帅,张玉文,杨志坚,郑金贵. 2017. 显齿蛇葡萄实时荧光定量PCR内参基因的筛选与验证[J]. 中草药,6(3):1192-1198. [Xu M,Yi H J,Zhao S,Zhang Y W,Yang Z J,Zheng J G. 2017. Selection and validation of reference genes for quantitative RT-PCR analysis in Ampelopsis grossedentata[J]. Chinese Traditional and Her-bal Drugs,6(3):1192-1198.]
严志祥,罗茜,张翼冠,赵军宁. 2019. 药用植物转录组研究现状与展望[J]. 中国药学杂志,54(7):513-520. [Yan Z X,Luo X,Zhang Y G,Zhao J N. 2019. A brief review on medicinal plants transcriptome research[J]. Chinese Pharmaceutical Journal,54(7):513-520.]
朱畇昊,董誠明,郑晓珂,冯卫生,刘孟奇,赵乐. 2017. 基于转录组测序的山茱萸次生代谢生物合成相关基因的挖掘[J]. 中国中药杂志,42(2):213-219.[Zhu Y H,Dong C M,Zheng X K,Feng W S,Liu M Q,Zhao L. 2017. Transcriptome analysis reveals genes involved in biosynthesis of secondary metabolism in Cornus officinalis[J]. China Journal of Chinese Materia Medica,42(2):213-219.]
朱畇昊,张梦佳,李璐,赵乐,董诚明. 2019. 夏枯草的转录组测序与次生代谢产物生物合成相关基因的挖掘[J]. 中草药,50(5):1220-1226. [Zhu Y H,Zhang M J,Li L,Zhao L,Dong C M. 2019. Transcriptome analysis of Prunella vulgaris and identification of putative genes involved in secondary metabolism biosynthesis[J]. Chinese Traditional and Herbal Drugs,50(5):1220-1226.]
邹福贤,许文,黄泽豪,张勋,陈抒云,林羽,徐伟. 2019. 金线莲转录组测序及其黄酮类合成相关基因分析[J]. 中国药科大学学报,50(1):66-74. [Zou F X,Xu W,Huang Z H,Zhang X,Chen S Y,Lin Y,Xu W. 2019. Analysis of transcriptome sequencing and related genes of flavonoid biosynthesis from Anoectochilus roxburghii[J]. Journal of China Pharmaceutical University,50(1):66-74.]
邹丽秋,王彩霞,匡雪君,李滢,孙超. 2016. 黄酮类化合物合成途径及合成生物学研究进展[J]. 中国中药杂志,41(22):4124-4128. [Zou L Q,Wang C X,Kuang X J,Li Y,Sun C. 2016. Advance in flavonoids biosynthetic pathway and synthetic biology[J]. China Journal of Chinese Materia Medica,41(22):4124-4128.]
Grabherr M G,Haas B J,Yassour M,Levin J Z,Thompson D A,Amit I,Adiconis X,Fan L,Raychowdhury R,Zeng Q,Chen Z,Mauceli E,Hacohen N,Gnirke A,Rhind N,di Palma F,Birren B W,Nusbaum C,Lindblad-Toh K,Friedman N,Regev A. 2011. Full-length transcriptome assembly from RNA-Seq data without are ference genome[J]. Nature Biotechnology,29(7),644-652.
Hichri I,Barrieu F,Bogs J,Kappel C,Delrot S,Lauvergeat V. 2011. Recent advances in the transcriptional regulation of the flavonoid biosynthetic pathway[J]. Journal of Experimental Botany,62(8):2465-2483.
Wilhelm B T,Landry J R. 2009. RNA-Seq-quantitative measurement of expression through massively parallel RNA-sequencing[J]. Methods,48(3):249-257.
Yang M,Zhou P N,Gui C,Da G Z,Gong L,Zhang X Q. 2019. Comparative transcriptome analysis of Ampelopsis megalophylla for identifying genes involved inflavonoid biosynthesis and accumulation during different seasons[J]. Molecules,24(7):1267-1282.
Zhang Y S,Yang W L. 2001. Study on functional constituents of Ampelopsis grossdentata[J]. Food Science,22(9):75-77.
Zhang Y S,Zhang Q Y,Wang B,Li L Y,Zhao Y Y. 2006. Chemical constituents from Ampelopsis grossedentata[J]. Journal of Chinese Pharmaceutical Sciences,15(4):211-214.
Zhou X J,Wang Y Y,Xu Y N,Yan R S,Zhao P,Liu W Z. 2015. De Novo characterization of flower bud transcriptomes and the development of EST-SSR markers for the endangered tree Tapiscia sinensis[J]. International Journal of Molecular Sciences,16(6):12855-12870.
(责任编辑 陈 燕)
收稿时间:2019-08-15
基金项目:国家科技支撑计划项目(2013BAD01B05);福建农林大学科技创新专项(KFA17424A)
作者简介:*为通讯作者,郑金贵(1949-),教授,主要从事地方特色作物种质资源挖掘与创新利用研究工作,E-mail:jgzheng@ fafu.edu.cn。许明(1978-),博士,主要从事药用植物生物技术研究工作,E-mail:xmfau@163.com
优 秀 青 年 学 者 论 坛
许明(1978-),博士,主要从事药用植物生物技术研究工作。先后主持或作为主要成员参与国家科技支撑计划项目“地方特色作物种质资源发掘与创新利用”、国家转基因重大专项“功能型转基因水稻新品种的培育”、农业农村部“948”计划项目“台湾优质农产品技术的引进、创新与推广”和福建省特色现代农业项目“特种作物新品种试验筛选与功能品质鉴定”等国家级、省部级科研项目10余项;在《Molecular Genetics and Genomics》《International Journal of Molecular Sciences》《Iranian Journal of Biotechno-logy》《中國农业科学》《南方农业学报》《西北植物学报》《中草药》《生物技术通报》等国内外核心期刊上发表学术论文20余篇;申报国家发明专利4项,获福建省科学技术奖三等奖1项。
猜你喜欢
中国医药导报(2017年9期)2017-05-11
中国中药杂志(2016年24期)2017-04-18
中国中药杂志(2017年4期)2017-03-28
中国中药杂志(2017年2期)2017-03-25
中国中药杂志(2017年3期)2017-03-20
科技创新导报(2016年28期)2017-03-14
中国中药杂志(2017年1期)2017-03-06
中国中药杂志(2016年22期)2017-02-13
江苏农业科学(2014年10期)2014-11-22
江苏农业科学(2014年1期)2014-07-18
