
拟南芥B3结构域蛋白质VALs互作转录因子的筛选
2020-11-02 08:48马瑞景张晴雯龚霞徐亚飞袁文雅
湖北大学学报(自然科学版) 2020年6期
马瑞景,张晴雯,龚霞,徐亚飞,袁文雅
(湖北大学生命科学学院, 湖北 武汉 430062)
0 引言
B3结构域蛋白是植物特有的一类转录因子家族,拟南芥中该家族成员至少含有87个成员,分为5个家族:Abscisic acid-insensitive 3 (ABI3),High level expression of sugar inducible (HSI),Auxin response fator (ARF),Related to ABI3/VP1 (RAV)和Reproductive meristem(REM)家族[1].在这5个家族中,关于ARF和RAV家族的研究较多,ABI3次之,近年来关于HSI家族的研究也日益增多.但是,对REM家族的研究较少,可能与家族成员繁多或存在冗余有关.不同的家族成员之间,B3结构域的DNA结合特异性也不同.ABI3和HSI家族成员的B3结构域可以识别Sph/RY元件的CATGCA序列[2],ARF家族成员的N端B3结构域可以识别TGTCTC序列[3],RAV家族成员的C端B3结构域可以识别CACCTG序列[4].由于其结合的基因序列不同,这暗示着不同家族可能参与生理过程也不同.
HSI家族成员又名为Viviparous 1/Abscisic acid insensitive-like(VAL)家族,是一类植物特异性的含B3结构域的转录抑制因子,除B3结构域外,还含有PHD (plant homeodomain)结构域、Zf-CW (zinc finger-cysteine and tryptophan residue-containing domain)结构域和EAR (ethylene-responsive element binding factor-associated amphiphilic repression) motif.VALs家族有3个成员,分别命名为VAL1,VAL2,VAL3,由于VAL3含有不完整的PHD结构域且苗期表达量很低,目前主要的研究是针对VAL1及VAL2.VAL1通过EAR motif与MEDIATOR CDK8模块的MED13的TRAP240结构域结合,通过Zf-CW motif与HDA6结合,使VAL1、MED13和HDA6形成复合体,直接结合到种子成熟基因的5’编码区,直接抑制拟南芥子叶中种子成熟基因的表达和脂肪酸的合成[5].Zf-CW结构域已经被证明是组蛋白标记的reader,能够特异地识别H3K4me2和H3K4me3[6];而PHD结构域也能与H3K4me3结合[7],这些证据表明VAL1可能通过调节组蛋白H3K9去乙酰化和H3K4甲基化来调节下游基因的表达.同时,也有研究证明VAL1和VAL2能够与FLC的CME元件(含Sph/RY motif)结合,招募PRC2复合体的LHP1到FLC染色质上使其组蛋白H3K4发生三甲基化,从而影响开花[8].
前期研究结果显示VALs蛋白主要通过B3结构域结合靶基因,并通过PHD、Zf-CW和EAR等结构域招募染色质修饰因子,抑制靶基因的表达.为了进一步揭示VALs蛋白抑制靶基因表达的分子机制,本研究拟通过酵母双杂技术筛选拟南芥的转录因子文库,获得一些与VALs蛋白直接互作的转录因子,为后续深入研究VALs蛋白发挥生物学功能的分子机制提供研究方向及基础.
1 材料与方法
1.1 实验材料拟南芥野生型材料Col和酵母菌株AH109均来自本实验室,拟南芥转录因子文库来自北京大学瞿礼嘉老师团队[9].
1.2 实验方法
1.2.1 VAL1与VAL2基因序列的分析及扩增 在TAIR网站(https://www.arabidopsis.org/)上查找VAL1和VAL2的CDS序列,设计引物;选取四叶期左右的嫩叶,用Trizolup (TransGen Biotech, Cat. No. ET111-01)提取拟南芥野生型Col的总RNA,之后用M-MLV Reverse Transcriptase (Invitrogen, Cat. No. 28025013) 反转录,以此为模板PCR扩增出基因序列,并测序验证.
1.2.2 Bait载体的构建 Gateway方法获得含VAL1、VAL2基因的Bait载体:在引物5’端加入attB序列,PCR扩增出带attB位点的基因序列(引物序列见表1),通过BP反应重组进入中间载体pDONR 201,再通过LR反应克隆进入最终载体pDEST 32形成Bait载体.BP反应体系:0.5 μL BP ClonaseTMII (Invitrogen)、0.5 μL pDONR201 (100 ng/μL左右)、3 μL attB-PCR product (50 ng/μL)、1 μL ddH2O;室温反应1 h,在反应体系中加0.5 μL 蛋白酶K于37 ℃水浴锅中孵育10 min,热激法转化大肠杆菌感受态细胞涂布于卡那霉素平板上,37 ℃倒置培养16 h.LR反应体系:0.5 μL LR ClonaseTMII (Invitrogen)、0.5 μL pDEST32 (100 ng/μL左右)、3 μL attL1 and attL2 Entry clone (50 ng/μL)、1 μL ddH2O;后续反应条件和转化方法操作同BP反应.

表1 本实验所用主要引物
1.2.3 Bait菌株的构建及自激活验证 构建的Bait质粒经测序验证后转入酵母AH109中:挑取新鲜的AH109单菌落摇培制成酵母感受态,加入50% PEG4000 240 μL、1 mol/L LiAc 34 μL和Bait质粒200 ng左右,混匀42 ℃水浴2 h离心取上清加水稀释涂布于亮氨酸缺陷型平板,28 ℃培养2~3 d得到转化菌株,阳检后获得Bait菌株.将两个Bait质粒与Prey空质粒pDEST22分别共转化进入酵母感受态AH109,涂布在SD/-Leu-Trp二缺培养基中,再分别挑取十几个共转化单菌落于200 μL ddH2O中,涡旋混匀后稀释10倍,用移液器吸取5~8 μL点在SD/-Leu-Trp二缺和SD/-Leu/-Trp /-His/-Ade四缺平板上,28 ℃倒置培养3~5 d比较共转化菌株在二缺和四缺培养基的生长状况,验证重组蛋白表达是否存在自激活现象.
1.2.4 拟南芥酵母转录因子文库的筛选 将Bait菌株摇菌培养至OD600约0.8左右,在2 mL 96孔深孔板中先加入200 μL YPDA培养基,然后加入30 μL Bait菌株,再加入30 μL事先融化的拟南芥酵母转录因子文库作为Prey菌株,盖上配套的硅胶盖,用保鲜膜封好,放入28 ℃摇床中50 r/min轻摇24 h左右;在每个孔中加入1 mL灭菌的ddH2O,然后吸取5~8 μL轻点在SD/-Leu-Trp二缺和SD/-Leu/-Trp/-His/-Ade四缺平板上,28 ℃倒置培养3~5 d观察实验结果.
2 结果与分析
2.1 VAL1与VAL2基因序列和蛋白质序列的分析VAL1基因序列号为AT2G30470,全长基因组序列为4 850 bp,选取AT2G30470.1转录本进行序列分析和后续载体构建,编码区有2 373 bp,包括12个外显子和11个内含子,编码氨基酸790aa;VAL2基因序列号为AT4G32010,全长基因组序列为5 976 bp,选取代表性转录本AT4G32010.1进行序列分析和后续实验,编码区有2 343 bp,也包括12个外显子和11个内含子,编码氨基酸780aa,全长基因组序列结构图如图1A和图1B.VAL1和VAL2的蛋白质长度相近,经过InterPro (http://www.ebi.ac.uk/interpro/)和Xfam (http://xfam.org)在线网址和相关文献鉴定两者的蛋白质结构,结果显示,VAL1和VAL2的结构也相同,都可以分为PHD结构域、B3结构域、CW结构域和EAR结构域[10],蛋白质结构图见图1C.PHD结构域是植物特有的一类含锌指结构的同源结构域,可以识别组蛋白修饰H3K4me3和K3K27me2以及H3K27me3,破坏VAL1的PHD结构域会导致种子相关基因富集的H3K4me3增加、K3K27me2和H3K27me3减少,使得这些基因在植物幼苗阶段异位表达影响植物的正常生长[11].B3结构域是DNA结合结构域,可以识别基于CATGCA序列的Sph/RY元件[2].CW结构域也能够识别组蛋白H3K4me2和H3K4me3,VAL2的CW结构域能够和组蛋白去乙酰化酶HDA19结合改变种子成熟基因的组蛋白乙酰化水平使其表达量下调[12].EAR结构域是能够招募如SIN3 (SWI-independent 3)和TPL (TOPLESS)等共抑制因子的一种抑制结构域,之后可以招募组蛋白去乙酰化酶复合体到靶基因上[13].
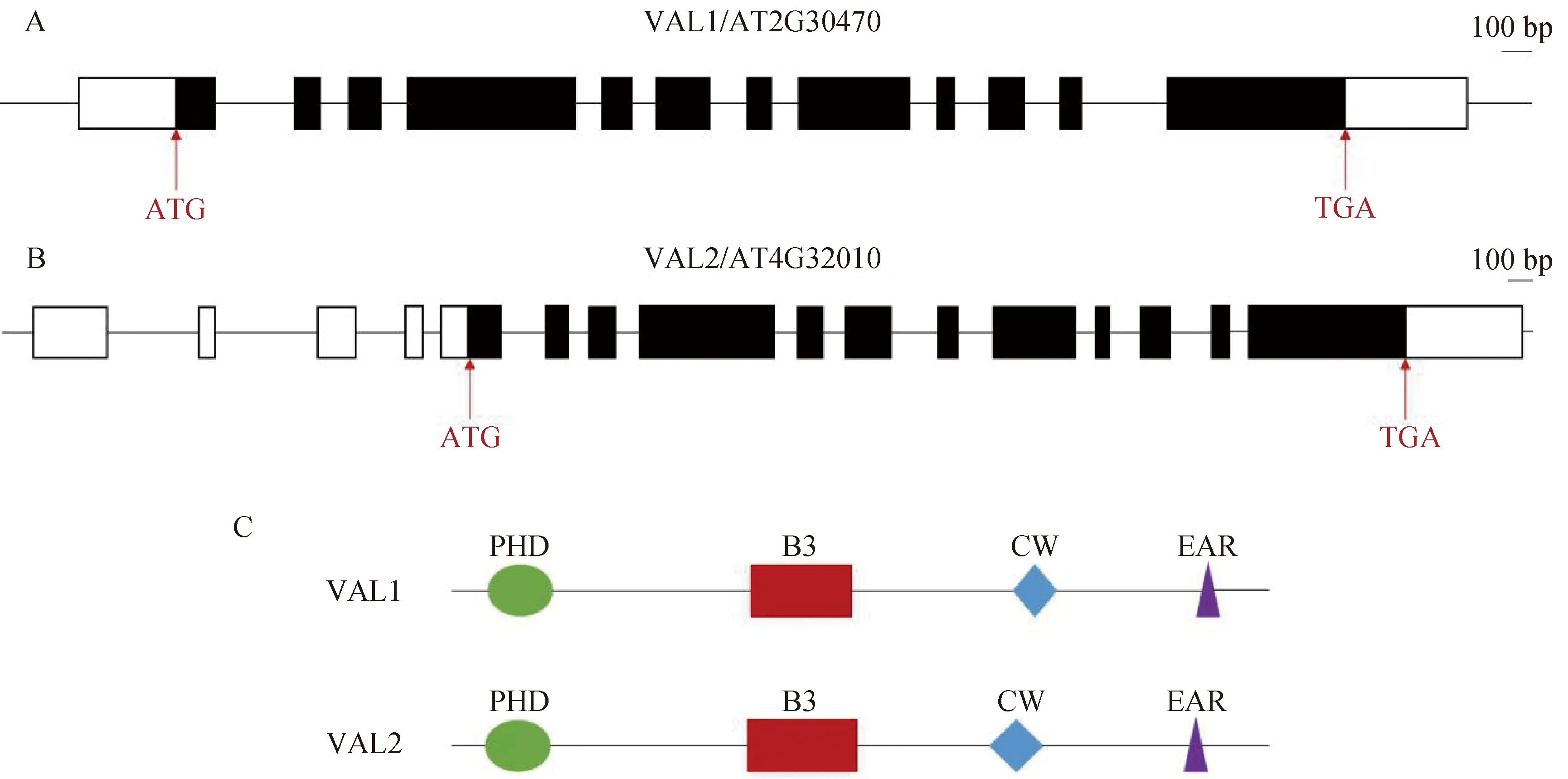
A和B分别是VAL1和VAL2的基因结构图,横线表示非编码区和内含子,白色方框表示5’UTR和3’UTR,黑色方框表示编码区及外显子;C为VAL1和VAL2的蛋白质结构示意图图1 VAL1和VAL2的基因结构图及蛋白质结构图

PCR扩增后产物的电泳胶图,泳道1:VAL1;泳道2:VAL2;泳道3:Marker图2 VAL1和VAL2的基因扩增图
2.2 VALs Bait载体的构建取拟南芥野生型Col的嫩叶用Trizol试剂提取总RNA,经过反转录酶反转录之后合成第一链cDNA.根据VALs基因序列设计引物,在引物5’端加上含attB位点的序列(上游引物5’端添加序列:GGGGACAAGTTTGTACAAAAAAGCAGGCTTC;下游引物5’端添加序列:GGGGACCACTTTGTACAAGAAAGCTGGGTT)合成引物.以反转录的cDNA为模板,用上述设计的引物进行PCR反应扩增出含attB位点的VAL1和VAL2的基因序列,基因扩增条带见图2,PCR产物送公司测序验证序列正确.
构建质粒载体有多种方法,本实验中所用到的拟南芥转录因子文库中Prey菌株所含的载体为pDEST 22,与之进行酵母双杂实验的配套Bait载体为pDEST 32.pDEST 22及pDEST 32载体为Gateway同源重组系统,所以将带attB位点的VAL1和VAL2基因经BP重组反应克隆到中间载体pDONR 201创建出entry clone:pDONR-201-VAL1和pDONR-201-VAL2,再经LR重组反应克隆到最终载体pDEST 32中成为expression clone:pDEST 32-VAL1和pDEST 32-VAL2,当中两次克隆均需挑阳性克隆送测序验证完全正确(Gateway技术原理示意见图3).

A为BP反应示意图; B为LR反应示意图图3 Gateway技术原理示意图[14]

W:Trp 色氨酸;L:Leu 亮氨酸;H:His 组氨酸;A:Ade 腺嘌呤图4 VAL1及VAL2的自激活验证

图5 VAL2与HD ZIP IV家族5个成员的酵母互作图
2.3 VALs Bait菌株的构建及自激活验证和毒性检测将测序验证完全正确的两个Bait质粒转入酵母AH109菌株,进行酵母菌落PCR筛选阳性克隆,将阳性克隆命名为Bait菌株B-VAL1和B-VAL2,配置50%甘油-80 ℃冻存菌株.在进行酵母双杂交之前,Bait菌株需进行自激活验证和毒性检测.自激活验证是为了确认Bait是否能自动激活菌株里的报告基因,可以采用将Bait菌株活化后直接涂布在加入X-α-Gal或Aureobasidin A抗生素的SD/-Leu单缺平板上,或者直接涂布在SD/-Leu-Trp二缺培养皿上.我们采用的是将Bait质粒和Prey空质粒pDEST22共转进入AH109直接涂布在二缺培养皿上,再将共转的菌株点在二缺和四缺平板上观察3~5 d后发现两个菌株在二缺上都能生长,在四缺上都不能生长,表明它们都不存在自激活现象(见图4).毒性检测是要检测该表达蛋白是否会影响酵母的生长,如果Bait菌株在单缺培养基的摇培过程中发现生长异常缓慢,说明该菌株有毒性效应,本实验中B-VAL1和B-VAL2无论在固体还是液体培养基中均能够正常生长.
2.4 拟南芥酵母转录因子文库的筛选酵母双杂实验的原理是转录因子Gal4含有一个DNA结合结构域(binding domain,BD)和一个激活结构域(active domain,AD),将其分别连在两个载体上,在这两个载体上分别接上Bait基因以及Prey基因;当两个载体转入一个菌株时,若这两个载体表达的蛋白质能够发生相互作用,AD和BD接近能够起到转录因子的作用激活菌株中的报告菌株,从而能够在营养缺陷型的培养基上正常生长.在本实验中,Bait菌株是AH109,转录因子文库作为Prey菌株是Y187,这两种菌株能够通过Mate接合生殖生成同时含Bait及Prey两种质粒的新的菌株来验证两个蛋白质是否能发生相互作用.拟南芥酵母转录因子文库共有14个96孔深孔板(P1~P14),包含1 344个Prey菌株,覆盖了拟南芥的大部分转录因子;我们分两批进行了两次重复的酵母双杂实验后筛选到了与VAL2能够发生相互作用的菌株有6个,分别为P7B7、P12H9、P12H10、P12H11、P13C9、P13C10.其中,两次重复都能鉴定到的基因有5个,分别为AT4G04890、AT1G05230、AT5G46880、AT1G73360、AT1G17920,基因名分别为PDF2、HDG2、HDG5、HDG11、HDG12(https://www.arabidopsis.org/),都为HD-ZIP IV家族的转录因子.将这5个基因对应的酵母菌株挑出来单独与B-VAL2(pDEST-32-VAL2)进行点对点Mate实验验证,结果显示VAL2确实与PDF2、HDG2、HDG5、HDG11及HDG12这5个蛋白存在相互作用(图5).另外,本次实验中并未筛选到能够与VAL1发生相互作用的蛋白质,但这并不意味着VAL1没有能与之互作的转录因子.
3 讨论与展望
3.1 酵母双杂交实验技术的优劣酵母双杂交实验是体外验证蛋白质互作的最经典、最快速的实验方法.当我们没有已转化目标检测蛋白的拟南芥转化植株,但是又想要先快速检测一下两种蛋白质是否能潜在地发生互作时,首选的是用时最短的酵母双杂实验.但是,除了酵母双杂交外,我们还需要利用其他的实验技术同时从体外体内两方面进行验证,比如BiFC、pull-down、Co-IP等技术.多种实验同时做出有互作的阳性结果才能最终确认这两个蛋白质存在相互作用.酵母双杂交可能存在假阴性或者假阳性,比如已发表的文献中做出某两个蛋白有相互作用时,但是我们并没有阳性结果,此时可能需要多重复几次实验或者进行其他实验验证;有时酵母双杂交实验有阳性结果,但是用更准确的体内实验验证并没有互作时,一方面可能是酵母实验存在假阳性,另一方面可能是这两个蛋白质只在体外互作,在植物体内并不发生互作.为了减少酵母假性实验结果,通常需要对Bait进行表达检测、自激活验证以及毒性检测,检测Bait重组蛋白是否能够成功表达,可以用GAL4 DNA-BD单克隆抗体或c-Myc单克隆抗体进行Western blot实验,通常实验中一般可以忽略这一步.我们后续将用其他技术继续验证VAL2与HD ZIP IV家族5个成员是否确实能在体外和体内互作参与某些生理过程,同时继续筛选与VAL1能发生互作的蛋白质.
3.2 HD-ZIP IV家族转录因子HD-ZIP指homeodomain-leucine zipper转录因子家族,HD-ZIP IV家族是该家族第四个亚族,共有16个成员,分别为HDG1-HDG12、GL2、ANL2、ATML1、PDF2.GUS启动子融合表达分析显示,HDG2、HDG5、HDG11、HDG12、ATML1、PDF2主要表达于茎分生组织和器官的表皮层,这表示HD-ZIP IV家族的许多成员可能参与调节表皮的基因表达.而这16个基因的突变体中,只有少数几个突变体有明显的表型性状.hdg2突变体的表皮毛(trichome)具有光滑的细胞壁[15],hdg11突变体的表皮毛分支增多且hdg11hdg12双突变体相对于hdg11单突变体表型明显增强[16].PDF2和ATML1的序列相似,并且都表达在茎尖分生组织的最外层L1层,它们的单突变体都没有明显的表型,但是双突变体在茎表皮细胞分化方面有着严重的缺陷,说明它们功能冗余且十分关键[17].同时,pdf2分别和hdg2、hdg5、hdg12的双突变体的花异常,表现为萼片花瓣和心皮雄蕊异常,这可能与花瓣和雄蕊识别基因APETALA 3 (AP3)的下调有关[18].这表明HD-ZIP IV家族不仅参与茎尖分生组织的发育,同时也参与花器官的形成,而VALs转录因子也参与幼苗早期生长和花期的调控[5, 8],说明VALs极有可能与HD-ZIP IV发生互作共同调控这两个阶段的发育.后续我们可以通过其他蛋白质互作技术继续验证VAL2与HD-ZIP IV家族的互作,同时寻找其突变体或采用CRISPR基因编辑技术敲除并观察双突变体或多突变体的表型,进而研究该互作在植物生长发育过程中起到的功能与意义.
猜你喜欢
亚热带农业研究(2022年1期)2022-08-08
湖北农业科学(2022年11期)2022-07-18
河北农业大学学报(2022年2期)2022-04-26
农业科技通讯(2021年1期)2021-03-06
园艺与种苗(2020年12期)2021-01-08
实用肿瘤学杂志(2020年4期)2020-12-08
中国农业科技导报(2020年3期)2020-03-15
山西农业科学(2020年2期)2020-02-29
飞碟探索(2015年9期)2015-11-05
红领巾·探索(2015年9期)2015-09-10
