
基于CRISPR/Cas9技术的β-地中海贫血和血友病基因治疗研究进展
2020-10-29 05:48:02鲍莉雯周一叶曾凡一
遗传 2020年10期
鲍莉雯,周一叶,曾凡一,
基于CRISPR/Cas9技术的β-地中海贫血和血友病基因治疗研究进展
鲍莉雯1,周一叶2,曾凡一1,2
1. 上海交通大学附属儿童医院,上海市儿童医院,上海交通大学医学遗传研究所,国家卫生健康委员会医学胚胎分子生物学重点实验室,上海市胚胎与生殖工程重点实验室,上海 200040 2. 上海交通大学基础医学院组织胚胎学与遗传发育学系,上海 200025
地中海贫血和血友病是由基因异常引发的常见的遗传性血液病,难以根治且可遗传给下一代,造成严重的家庭和社会负担。基因治疗的出现为遗传性疾病提供了新的治疗方案,但自1990年第1项基因治疗临床试验被批准以来,30年间基因治疗的发展并不乐观。随着基因编辑技术的发展,尤其具有编辑效率高、操作简单、成本低等优势的第三代基因编辑技术CRISPR/Cas9 (clustered regularly interspaced short palindromic repeats/ CRISPR-associated protein 9)的发展,基因编辑介导的基因治疗越来越受到关注,有望根治地中海贫血和血友病等遗传性血液病。本文综述了近6年(2014~2020年)基于CRISPR/Cas9技术的β-地中海贫血和血友病基因治疗基础研究进展,总结了基于CRISPR/Cas9技术的基因治疗临床试验概况,并对CRISPR/Cas9技术用于基因治疗存在的问题和可能的解决方案进行探讨,以期为基于CRISPR/Cas9技术的遗传性血液病基因治疗相关研究提供参考。
CRISPR/Cas9;基因治疗;遗传性血液病;临床试验
遗传性血液病是由基因异常引发的血液疾病,研究较为透彻的有地中海贫血(地贫)和血友病。在红细胞中,血红蛋白负责运输氧和二氧化碳,由两条类α珠蛋白链(α链或ξ链)和两条类β珠蛋白链(β链、γ链或ε链)各结合一个血红素组成[1]。珠蛋白基因突变会引起血红蛋白病,包括血红蛋白变异体和地贫,前者受累于血红蛋白结构异常,后者则由于α链或类β链合成数量减少/缺失,致使另一条珠蛋白链含量相对过剩并沉积于红细胞膜,进而引发溶血,严重可导致胎儿流产和患儿死亡[1,2]。根据减少/缺失的珠蛋白链的不同,地贫可分为α-地中海贫血(α-地贫,α链合成减少/缺失)、β-地中海贫血(β-地贫,β链合成减少/缺失)等[3,4]。在我国广西壮族自治区α-地贫发病率高达14.95%,在广州β-地贫发病率高达2.21%[3]。
血友病是由凝血因子基因突变导致相应凝血因子严重缺乏而引起的出血性疾病,其中血友病A和血友病B最常见且均为X连锁隐性遗传病,分别由于凝血因子VIII基因(factor VIII,)和凝血因子IX基因(factor IX,)突变而引起[5](表1)。
对于上述遗传性血液病,临床上主要采取长期规范输血、成分输血等方法进行对症治疗[1]。这些短效的对症疗法无法根治遗传性血液病,且长期反复输血会导致铁沉积,还可能引发感染和过敏反应,甚至导致死亡。少部分患者可通过异体骨髓或造血干细胞移植获得长效/永久的治疗效果,但存在供体缺乏和免疫排斥等问题,且费用昂贵。随着现代基因编辑技术的进步,基因治疗成为最具前景的治疗方案,有望根治遗传性血液病。本文从基础研究和临床研究两方面对基于CRISPR/Cas9技术的β-地贫和血友病的基因治疗研究进展进行综述,以期为基于CRISPR/Cas9技术的遗传性血液病基因治疗相关研究提供参考。

表1 常见遗传性血液病
1 遗传性血液病基因治疗概况
1990年开展的针对遗传性血液病——重症联合免疫缺陷的基因治疗试验被认为是首次成功的基因治疗临床试验。该研究通过基因增补的方式,利用逆转录病毒将正常的腺苷脱氨酶(adenosine deaminase,)基因体外导入患者T细胞,并随机插入基因组,改造后的T细胞输回患者体内用于治疗[6]。但逆转录病毒介导的基因转导存在安全问题,且淋巴细胞寿命短,不利于发挥长效的治疗效果。此后,多种病毒和非病毒载体被开发,以提高安全性。改造的细胞也能转为无限增殖并分化的造血干细胞和祖细胞(hematopoietic stem and progenitor cell, HSPC),以实现长效治疗。除了体外改造患者细胞的体外途径,基因治疗还可通过体内途径实现[7],即直接将载有目的基因的载体通过静脉输注、肌肉注射等方式输入患者体内,目前血友病的基因治疗主要采取体内途径,比如,通过门静脉输注将载有凝血因子VIII或凝血因子IX基因的肝趋向性腺相关病毒载体(adeno-associated virus, AAV)注入患者体内实现基因治疗[8,9](图1)。但体内途径的基因治疗过程难以控制,出于安全性考虑,以造血干细胞为靶细胞的基因治疗主要采取体外途径。
对于β-地贫还可通过调控基因表达的方式进行治疗。上海医学遗传研究所利用羟基脲治疗β+地贫患者(患者能部分合成正常β珠蛋白链),首次发现低剂量的羟基脲可增加正常β链的合成,并显著改善患者表型[10]。此外,由于β-地贫患者体内α链相对过剩,减少α链合成可缓解疾病症状,如利用RNA干扰(RNA interference, RNAi)技术下调α-珠蛋白基因()表达可缓解β-地贫小鼠模型的贫血症状[11,12]。γ链可在功能上代偿β链的缺失或异常,重激活γ-珠蛋白基因()表达可改善β-地贫和镰状细胞贫血症状[13,14]。
理想的基因治疗是突变基因的原位修复,目前主要通过基因编辑技术实现。经过第一代锌指核酸酶(zinc finger nuclease, ZFN)、第二代转录因子样效应物核酸酶(transcription activator-like effector nuclease, TALEN),基因编辑技术已发展至第三代——CRISPR/Cas9,进一步又发展出单碱基编辑技术(base editing, BE)[15,16]。虽然利用病毒载体增补基因是目前基因治疗临床试验的主流,但基于基因编辑技术的基因治疗临床试验项目正逐年增多,尤其CRISPR/Cas9技术介导的基因治疗临床试验已超过ZFN和TALEN相关临床试验总数。在疾病选择方面,因β-地贫和血友病等致病机制清楚,均为单基因病,且主要突变类型为点突变或移码突变,易于实施基因治疗,成为遗传性血液病基因治疗研究的主要研究对象。

图1 CRISPR/Cas9用于遗传性血液病基因治疗的研究
sgRNA:向导RNA;RNP:核糖核蛋白复合体;HSPC:造血干细胞或祖细胞;iPSC:诱导多能干细胞。
2 基于CRISPR/Cas9技术的遗传性血液病基因治疗策略
CRISPR/Cas9系统由向导RNA (single-guide RNA, sgRNA)和Cas9蛋白组成[17~19],与ZFN和TALEN技术类似,CRISPR/Cas9技术同样通过诱导DNA双链断裂,诱发同源重组修复(homology- directed repair, HDR)和/或非同源末端连接(non- homologous end joining, NHEJ)等DNA损伤修复过程,实现基因编辑[19~21](图2)。其中同源重组修复需要同源模版,DNA双链断裂按模版精确修复,只在S期晚期和G2期姐妹染色单体存在时起作用;非同源末端连接无需同源模版,无法精确修复DNA双链断裂,修复时在双链断裂处造成核苷酸插入或缺失(insertions and/or deletions, indel),可删除突变位点,或改变关键DNA序列引起基因表达改变,在整个细胞周期均可发生[22,23](表2)。两种途径的选择主要由同源模版和细胞所处细胞周期决定,难以人为调控。
基于CRISPR/Cas9系统,单碱基编辑技术被开发出来。单碱基编辑技术通过在无核酸酶活性的Cas9蛋白(dCas9)或只有切割DNA单链活性的Cas9蛋白(Cas9-nickase, Cas9n)上融合胞嘧啶脱氨酶或腺嘌呤脱氨酶,在不剪切DNA双链的情况下实现C>T (G>A)或A>G (T>C)的单碱基编辑,不依赖同源重组修复和非同源末端连接等DNA损伤修复过程,无需同源模版便可实现点突变的精确修复,且编辑效率最高可达100%[24~26]。
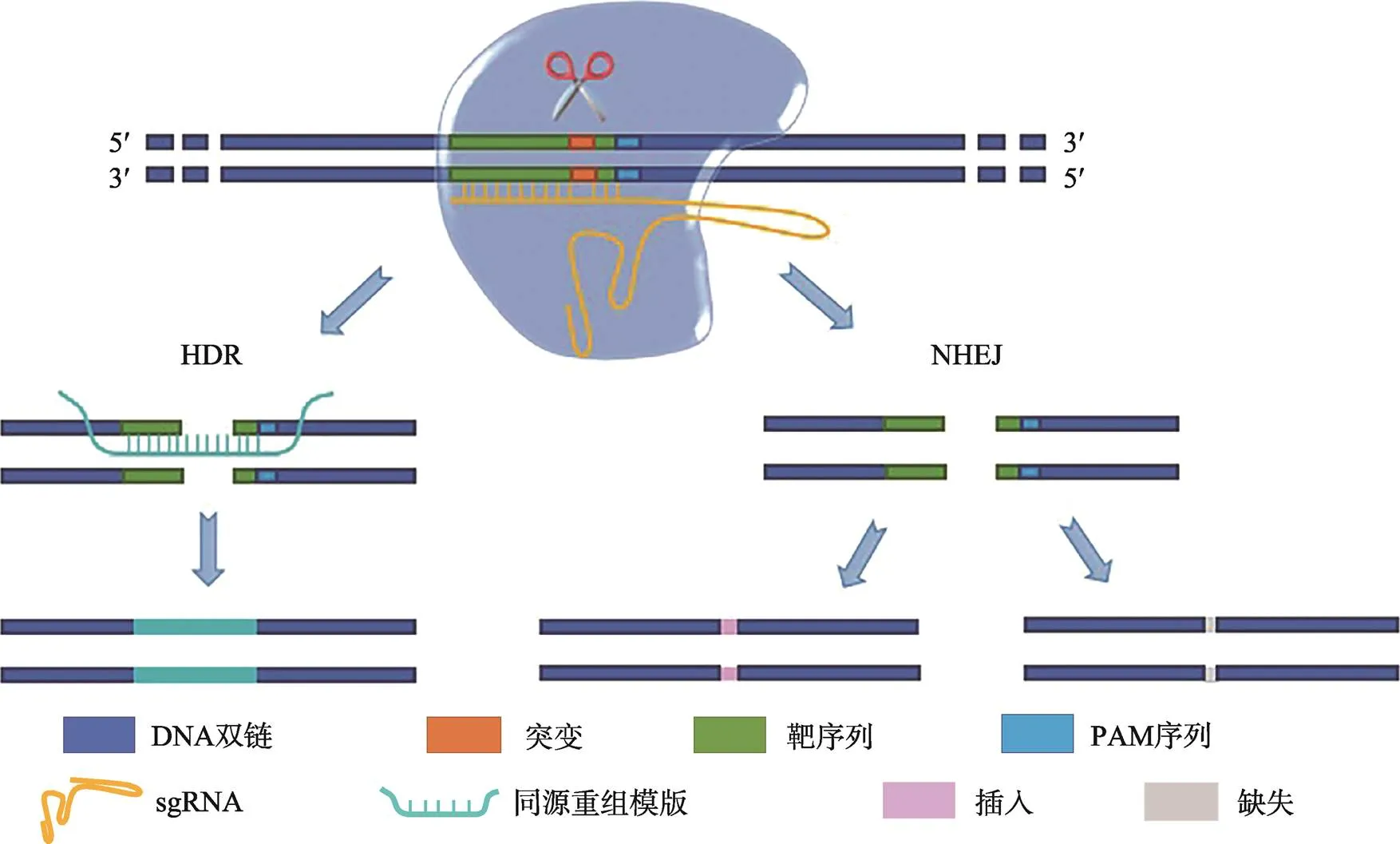
图2 CRISPR/Cas9基因编辑原理
HDR:同源重组修复途径;NHEJ:非同源末端连接途径。

表2 两种DNA损伤修复途径
2.1 原位修复突变基因
2.1.1 CRISPR/Cas9介导的外显子点突变/移码突变的修复
外显子点突变/移码突变可导致蛋白质结构或合成数量异常,引发疾病。如我国常见的两种引起β-地贫的β-珠蛋白基因()突变CD41/42(-TCTT)和CD17(A>T)[27]。由于外显子编码蛋白质,外显子突变需精确修复才可恢复基因功能,因此外显子突变必须依赖同源重组修复途径精确修复,但在造血干细胞和诱导多能干细胞(induced pluripotent stem cell, iPSC)中,同源重组效率低且不稳定(0.045%~ 57%) (表3)[15,28~32]。靶细胞类型、靶细胞所处细胞周期、Cas9蛋白、sgRNA、同源模版以及CRISPR/Cas9系统的递送方式等的差异是造成同源重组效率低且不稳定的可能原因,优化这些参数或可提升同源重组效率。此外,2018年报道的研究利用CRISPR/Cas9技术修复HbE/β-地贫患者(β-珠蛋白等位基因一个带有26号密码子G>A点突变,产生异常β链进而产生异常血红蛋白HbE;另一个带有CD41/42-TCTT缺失,导致β链缺失)来源iPSC中的26号密码子G>A点突变,发现正确修复突变的iPSC经红系分化后β-珠蛋白基因mRNA和蛋白无显著提升,作者分析可能由于β-珠蛋白基因表达所需的转录因子KLF1和BCL11A的表达水平低所致,但遗憾的是该研究未分析修复后细胞产生的β-珠蛋白是否为正常β-珠蛋白[32]。这提示,对基因组的修复可能无法实现转录水平和蛋白水平的修复,导致无效的基因治疗,因此突变修复后的细胞是否能产生正常的转录本并翻译出足够的功能正常的蛋白质也是CRISPR/Cas9用于基因治疗需要考虑的问题。

表3 CRISPR/Cas9原位修复突变基因的研究
HDRa:同源重组修复;NHEJb:非同源末端连接;NAHRc:非等位同源重组;HiFi Cas9d:高保真Cas9。
2.1.2 CRISPR/Cas9介导的内含子点突变的修复
内含子点突变可能导致mRNA剪接异常,影响蛋白质翻译,从而引发疾病。例如,导致β-地贫的β-珠蛋白基因IVS2-654 (C>T)突变,造成β-珠蛋白mRNA前体剪接异常,产生的异常mRNA (254 bp)较正常mRNA (181 bp)多出73 bp的内含子序列,进而影响β-珠蛋白的合成[11]。内含子不编码蛋白,内含子突变只要不影响mRNA前体的剪接,便不影响基因功能,因此无需精确修复,同源重组修复和非同源末端连接两种DNA双链断裂修复途径都可恢复内含子突变基因的功能[33~35]。通过非同源末端连接途径修复内含子突变附近的DNA双链断裂可高效删除内含子突变(效率可达98%) (表3),恢复基因功能[34,35]。但非同源末端连接途径产生的插入或缺失具有异质性,这些插入或缺失的产生是否会引入新的突变,造成基因表达和蛋白功能的异常,值得进一步研究。
2.1.3 CRISPR/Cas9介导的内含子倒位的修复
内含子倒位是引起重型血友病A的常见突变类型,约50%重型血友病A患者因凝血因子VIII基因1号或22号内含子倒位破坏了基因编码区,进而导致凝血因子VIII转录本的缺失,无法合成凝血因子VIII而致病[36]。非等位同源重组(non-allelic homologous recombination, NAHR)是导致凝血因子VIII基因内含子倒位突变的原因[36]。非等位同源重组由基因组中与同源模版序列类似的非同源序列(非同源模版)介导,是引起DNA重排的关键机制之一[37,38]。凝血因子VIII基因含有的两个1号内含子同系物(int1h-1、int1h-2)和3个22号内含子同系物(int22h-1、int22h-2、int22h-3)可作为非同源模版诱发凝血因子VIII基因上的DNA双链断裂经非等位同源重组途径错误修复,产生内含子倒位[36]。
研究表明模拟这一过程可修复倒位突变[39]。通过在倒位片段的两端设计向导RNA,引导Cas9切割倒位片段两端,诱发非等位同源重组,使倒位片段再次发生倒位,实现患者iPSC中内含子倒位的修复,基因修复的细胞分化后凝血因子VIII基因表达量由0上升至正常水平的50%~120%,但修复效率只有3.7%[36]。这可能由于细胞中存在一系列抑制非等位同源重组的调控机制[40](表3)。
2.1.4 单碱基编辑技术介导的原位修复
对于单碱基突变引起的遗传性血液病,除了上述利用CRISPR/Cas9技术进行原位修复,还可通过单碱基编辑技术实现突变的精确修复。对β-珠蛋白基因上–28(A>G)突变进行单碱基编辑,结果表明患者来源的造血干细胞中有36.4%的突变实现精确修复(C>T),56.4%的突变被错误编辑(C>G/A),3.6%的突变未被编辑[41]。说明在患者造血干细胞中单碱基编辑技术介导的精确修复效率有待提升。此外单碱基编辑技术还存在脱靶率高的问题,研究表明单碱基编辑系统可造成基因组水平和转录组水平的大范围脱靶[24]。在HEK293T细胞系中单碱基编辑系统BE3可导致近100%的RNA单核苷酸变异[42]。向脱氨酶中引入点突变可能是减少脱靶的有效方法[42]。
2.2 调控基因表达
α链和β链数量不平衡是导致地贫的直接原因,β-地贫患者的β链合成减少或缺失导致α链相对过剩,减少α链合成可缓解疾病症状[12,43]。对α-珠蛋白的顺式调控元件进行基因编辑,可调控α-珠蛋白的表达量。研究表明利用CRISPR/Cas9敲除β-地贫患者造血干细胞中α-珠蛋白基因增强子MCS-R2的核心元件,可降低α链的合成水平[44]。
γ链是胎儿血红蛋白HbF (fetal hemoglobin, α2γ2)的组成部分,在胎儿时期高表达[43]。出生后,γ链逐渐被β链取代,人体内主要血红蛋白转变为HbA (adult hemoglobin, α2β2)[43](图3)。重激活γ-珠蛋白基因,产生γ链与α链结合形成HbF,可代替HbA发挥功能,缓解α链相对过剩导致的地贫症状和β链结构异常导致的镰状细胞贫血症状[45~48]。
γ-珠蛋白基因表达受转录因子、启动子、增强子等多种元件调控。其中转录抑制因子BCL11A等是成人红系细胞中γ-珠蛋白基因低表达的重要影响因素[46,49]。BCL11A可以结合γ-珠蛋白基因的近端启动子,并抑制γ-珠蛋白基因表达。由于BCL11A在淋巴细胞发育中有重要调控作用,敲除造血干细胞中的虽可显著提升其分化的红系细胞的γ-珠蛋白基因的表达水平,但会影响淋巴细胞的成熟[50,51]。进一步研究发现存在一个红系特异增强子,在造血干细胞中敲除该增强子的DNA酶I超敏位点(DNase I hypersensitive site, DHS) +58可特异下调红系细胞中的表达,而不影响其他类型细胞,且效果与直接敲除相似[45,49]。利用CRISPR/Cas9在β-地贫和镰状细胞贫血患者的造血干细胞和祖细胞中敲除此位点,其分化后的红系细胞可产生治疗水平的胎儿血红蛋白[45](表4)。基于这一思路,相关临床试验已展开(表5)。
此外,重激活γ-珠蛋白基因还可采取其他方法,如破坏γ-珠蛋白基因近端启动子上转录抑制因子BCL11A或ZBTB7A的结合位点[46],其中BCL11A结合位点(TGACCA:启动子的−114至−119)是基因编辑的理想靶点,在β-地贫患者的造血干细胞和祖细胞中利用CRISPR/Cas9破坏该位点,平均编辑效率可达85%,编辑后的造血干细胞和祖细胞经红系分化,γ-珠蛋白的mRNA可达α-珠蛋白的126%,或利用单碱基编辑器(hAPOBEC3A-Cas9n, hA3A-BE3)破坏该位点,可使γ-珠蛋白mRNA与α-珠蛋白mRNA的比值由~6.8%升至~44.2%[52];模拟引起遗传性胎儿血红蛋白持续存在综合症的–113A>G突变,在γ-珠蛋白基因近端启动子上创造新的转录激活因子GATA1结合位点[47];敲除β-珠蛋白基因簇上可能的γ-珠蛋白基因抑制因子区域[48];下调血红素调节抑制因子(heme-regulated inhibitor, HRI)进而引起表达下调[53]等(表4)。
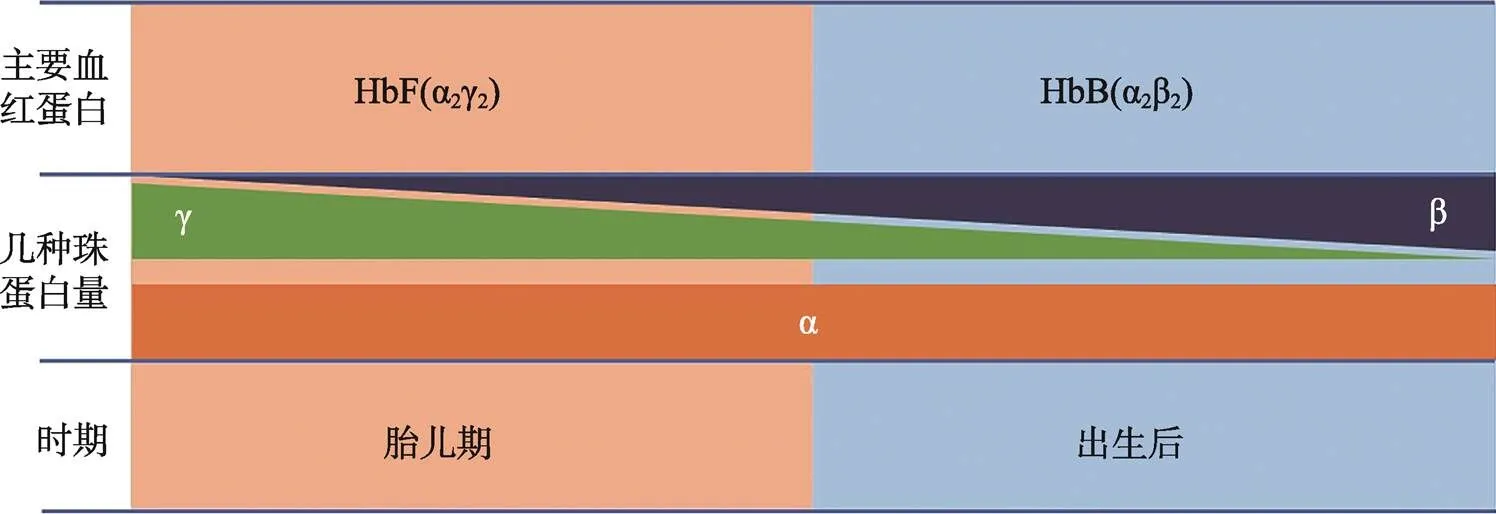
图3 血红蛋白演变示意图
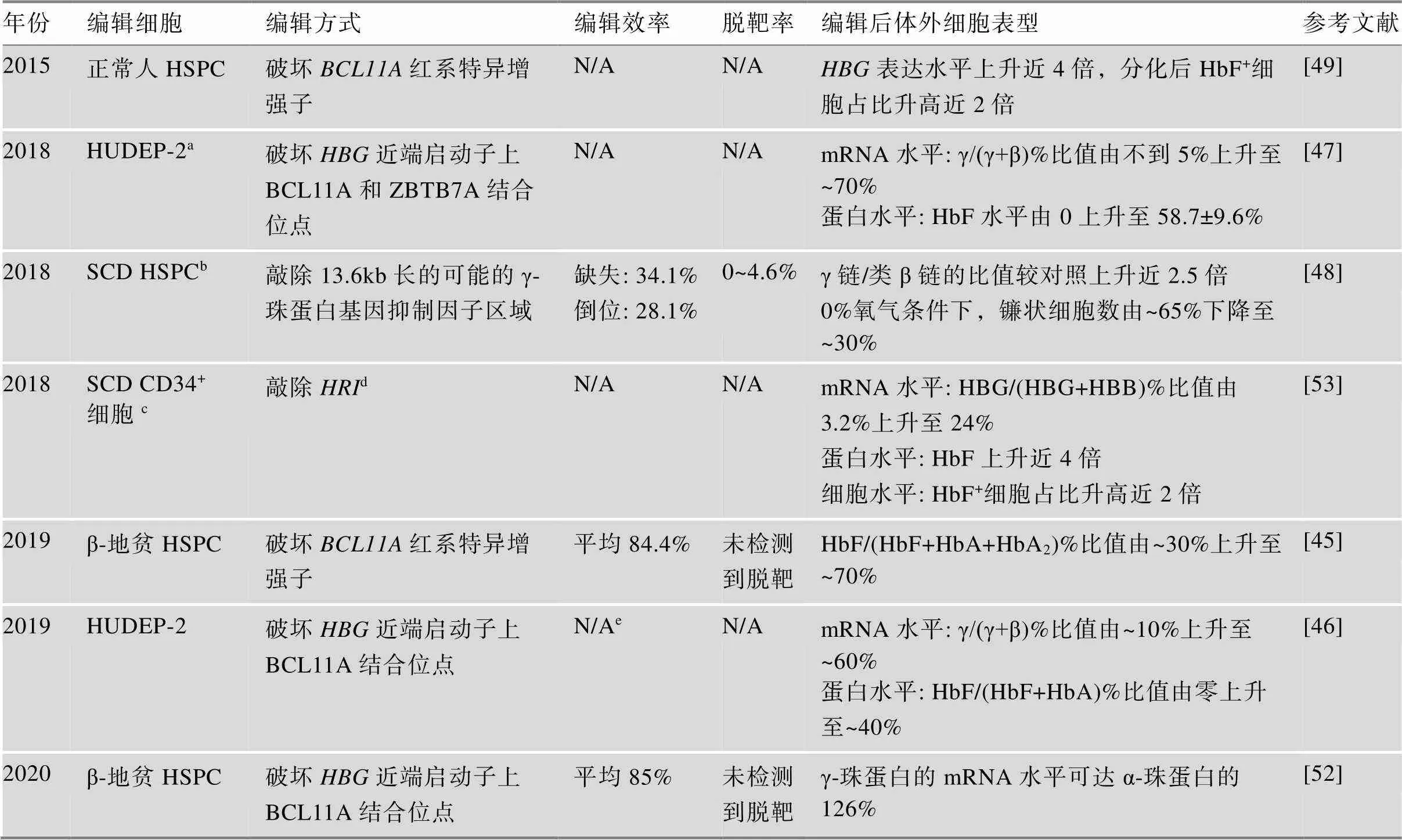
表4 基因表达调控: CRISPR/Cas9体外重激活HBG的基础研究
HUDER-2a:永生化人CD34+HSPC来源的红细胞系;SCD HSPCb:镰状细胞贫血症患者造血干细胞和祖细胞;SCD CD34+细胞c:镰状细胞贫血症患者CD34+细胞;d:血红素调节抑制因子基因;N/Ae:无数据。
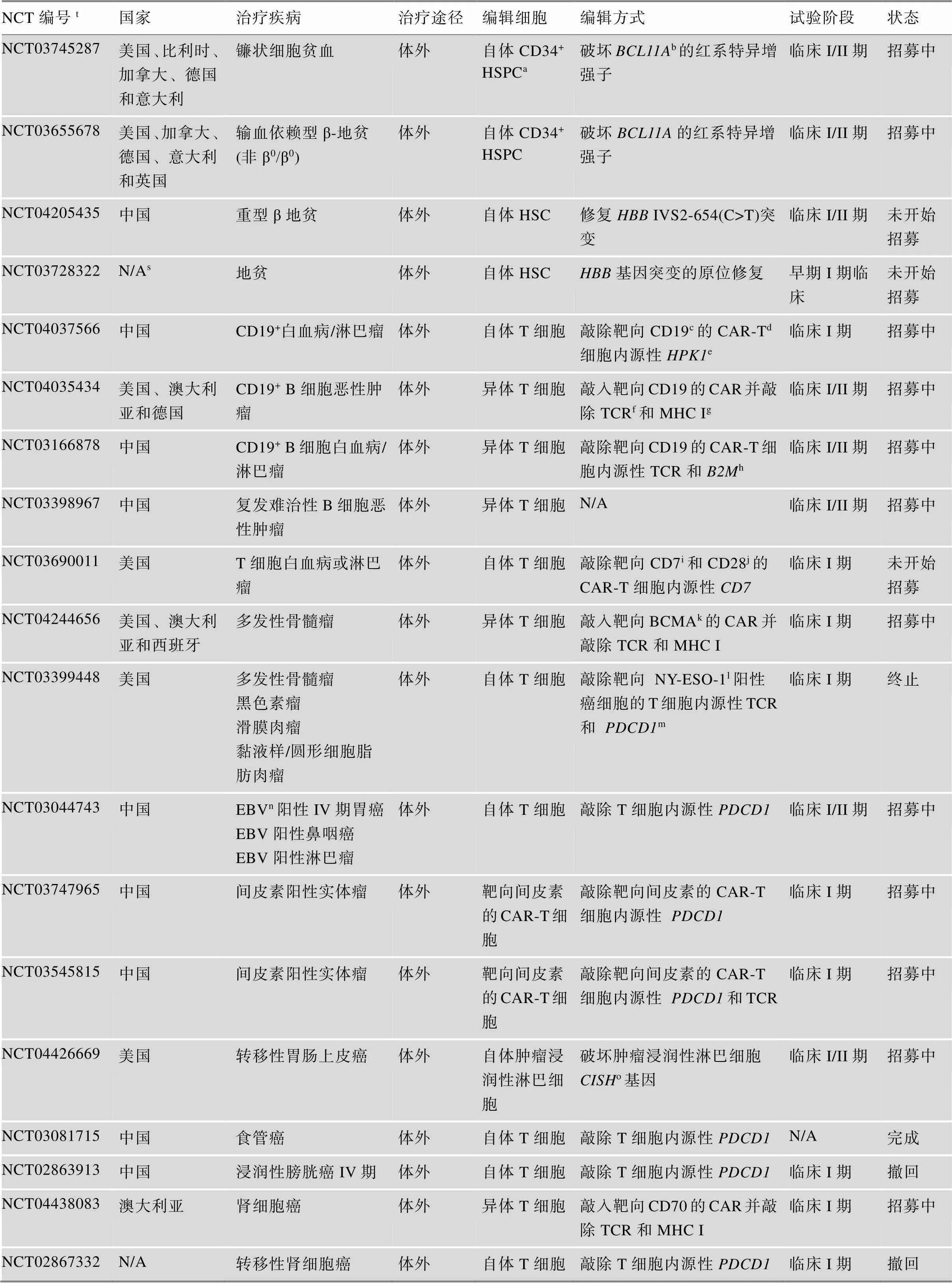
表5 基于CRISPR/Cas9技术的基因治疗临床试验

续表5
HSPCa:造血干细胞和祖细胞;b:B细胞淋巴瘤因子11A;CD19c:B细胞表面抗原;CAR-Td:嵌合抗原受体T细胞;e:丝裂原活化蛋白激酶激酶激酶1基因;TCRf:T细胞受体;MHC Ig:主要组织相容性复合体;h:β-2-微球蛋白基因;CD7i:白细胞分化抗原之一;CD28j:白细胞分化抗原之一;BCMAk:肿瘤坏死因子受体超家族成员17;NY-ESO-1l:癌症–睾丸抗原1;m:程序性细胞死亡蛋白1基因;EVBn:Epstein-Barr virus,EB病毒;o:细胞因子诱导含SH2蛋白基因;p:C-C基序趋化因子受体5基因;q:赖氨酸甲基转移酶2D基因;IVS26r:中心体蛋白290 26号内含子;N/As:无数据;NCT编号t:每一NCT编号附有相应网页链接,点击链接或在https://clinicaltrials.gov上搜索相应NCT编号可见各研究详情。
3 CRISPR/Cas9治疗策略与其他基因疗法的比较
对于遗传性血液病,目前利用病毒载体增补基因的基因治疗方法仍是临床试验的主流,尤其安全性更高、包装能力更强的慢病毒载体应用最为广泛。2018年报道的临床试验利用慢病毒载体将β-珠蛋白基因导入患者造血干细胞,对22例输血依赖型地贫患者进行基因治疗,其中12例非β0/β0基因型患者停止接受红细胞输入,另9例β0/β0基因型或双IVS1-110突变患者年输血量中位值降低了73%[54],证实了通过增补基因治疗β-地贫的可行性。但基因的随机整合存在插入突变等安全问题[55],因此与整合型病毒介导的基因增补相比,CRISPR/Cas9技术能直接靶向突变位点,目的性强。
采用非整合型病毒可降低随机整合引起的安全问题,如腺相关病毒,一般在胞内以附加体形式存在,基本不整合到基因组中,但其导入基因的长期表达效果不理想[56]。将腺相关病毒载体注射到体内增补凝血因子VIII或凝血因子IX基因是血友病主要的基因治疗方法[9]。对于血友病B,2017年报道的临床试验中凝血因子IX平均促凝活性水平可达33.7±18.5%,持续时间超1年[9,57];另一项研究中凝血因子IX表达时间可达8年甚至更久[9]。在血友病A基因治疗临床试验中,凝血因子VIII促凝活性水平可达52.3%,表达时间可达3年[9]。但随着细胞分裂,非整合型的腺相关病毒载体可能被丢失[56]。理想状态下,CRISPR/Cas9系统只需短暂存在于待编辑的细胞中便可实现永久的靶向基因修复。有动物实验通过颞静脉注射,将载有CRISPR/Cas9基因编辑系统的腺相关病毒载体注入凝血因子IX敲除的小鼠模型中,以实现人凝血因子IX的敲入[58]。治疗后,小鼠体内人凝血因子IX活性达正常水平的120%~ 160%,并至少可持续32周[58]。目前尚无基于CRISPR/ Cas9技术的血友病基因治疗相关临床试验。
与其他基因编辑技术相比CRISPR/Cas9技术主要胜在应用面广、易操作、效率高。不同于ZFN和TALEN需通过合成特殊蛋白识别靶DNA,CRISPR/ Cas9只需借助一段22nt的与DNA碱基互补配对的向导RNA,避免了复杂且昂贵的蛋白设计合成过程,且向导RNA设计合成灵活简便,CRISPR/Cas9系统装配简单,使得CRISPR/Cas9技术迅速得到广泛应用,包括用于基因治疗[59](表6)。
单碱基编辑系统对靶位点的识别机制与CRISPR/ Cas9相同,不同的是,CRISPR/Cas9介导的基因编辑依赖同源重组修复和非同源末端连接等DNA损伤修复途径对DNA双链断裂的修复[16]。同源重组修复虽能精确修复突变但效率低,非同源末端连接会造成核苷酸插入或缺失,可能引入新的突变[16]。单碱基编辑技术不造成DNA双链断裂,其介导的基因编辑不依赖同源重组修复和非同源末端连接等DNA损伤修复过程,不会造成核苷酸插入或缺失,且对单个碱基的精确编辑效率高,可达100%[16,25,26]。但单碱基编辑系统只能实现单个碱基的编辑,而CRISPR/Cas9除可实现单个碱基的编辑外,还可实现DNA大片段的插入或敲除。
4 基于CRISPR/Cas9技术的基因治疗临床研究
2016年中国开展了世界首个基于CRISPR/Cas9技术针对转移性非小细胞肺癌的基因治疗临床试验[60]。截止至2020年6月,基于CRISPR/Cas9技术的基因治疗临床试验共26项(表5),涉及遗传性血液病、血液肿瘤、实体瘤、感染性疾病和罕见病。对于血液肿瘤和实体瘤,CRISPR/Cas9主要用于编辑嵌合抗原受体(chimeric antigen receptor, CAR) T细胞(CAR-T cell)和患者自体T细胞,以提高治疗的安全性和有效性。2020年5月上述首个基于CRISPR/ Cas9技术的基因治疗临床试验(NCT02793856)公布了最新结果[61]。该研究利用CRISPR/Cas9技术体外破坏患者自体T细胞的基因,以激活T细胞杀伤癌细胞,二代测序结果表明编辑效率中位数为5.81% (范围0.42%~24.85%),脱靶率中位数为0.05% (范围0~0.25%)[61]。12名晚期非小细胞肺癌患者接受基因编辑T细胞治疗,无患者获得病情的部分缓解,2名患者获得病情稳定,未见严重不良反应[61]。另一项临床试验针对癌症–睾丸抗原NY-ESO-1和/或LAGE-1阳性肿瘤(NCT03399448),通过慢病毒载体将靶向NY-ESO-1和LAGE-1的T细胞抗原受体(T cell receptor, TCR)编码序列导入患者自体T细胞,使之靶向NY-ESO-1和/或LAGE-1阳性肿瘤细胞,同时利用CRISPR/Cas9技术敲除T细胞内源TCR和,以增强T细胞对肿瘤的杀伤力[62]。2名难治性晚期骨髓瘤患者和1名难治性静态肉瘤患者接受基因编辑T细胞输注后,2名患者获得病情稳定,未见细胞因子释放综合征或细胞输注引起的明显副作用[62]。感染性疾病包括人乳头瘤病毒感染和艾滋病。对于前者,研究利用CRISPR/Cas9靶向破坏人乳头瘤病毒的E6和E7基因,使病毒丧失感染能力。相反,艾滋病的治疗试图利用CRISPR/Cas9靶向破坏编码人类免疫缺陷病毒1型(human immunodeficiency virus-1, HIV-1)主要受体蛋白的基因,使HIV-1无法感染淋巴细胞。2019年该临床试验(NCT03164135)公布研究结果,1名合并有急性淋巴细胞白血病的艾滋病患者接受基因敲除的异体CD34+造血干细胞和祖细胞输注后急性淋巴白血病得到完全缓解,携带突变的造血干细胞和祖细胞在患者体内可长期存活达19个月,但不足以治愈HIV-1感染导致的艾滋病,研究未见基因编辑相关的不良反应[63]。上述临床研究结果显示了基于CRISPR/Cas9技术的基因治疗的可行性及安全性,但有效性有待提升。罕见病有歌舞伎综合征1和Leber先天性黑蒙10型(Leber congenital amaurosis type 10, LCA-10),其中针对Leber先天性黑蒙10型的临床试验是第一项也是目前唯一一项CRISPR/Cas9介导的体内途径的基因治疗临床试验[64]。

表6 基因编辑技术对比
总体来看,基于CRISPR/Cas9技术的基因治疗临床试验以体外途径为主,主要集中于癌肿治疗(20项),可能由于癌肿往往严重威胁患者生命,癌肿的治疗是目前临床上迫切需要解决的问题。从疾病发生系统来说,则主要集中于血液系统疾病(13项),这可能得益于造血干细胞移植的成功和CAR-T细胞疗法在血液肿瘤中取得了较好的临床效果[65]。在遗传性血液病方面,目前临床研究数相对较少(4项),可能由于遗传性血液病目前已有疗法有一定效果,且CRISPR/Cas9系统存在的缺陷暂未很好解决,目前相关临床研究结果尚未发布。
5 结语和展望
CRISPR/Cas9技术走向临床展现了其用于基因治疗的良好前景,但其存在的问题仍不容忽视。(1)安全性问题。研究表明CRISPR/Cas9技术存在严重脱靶效应,脱靶率可达靶向编辑效率的5.6%~125%[66]。最新研究报道Cas9蛋白可能多次切割DNA,造成百万碱基级的染色体末端大片段缺失[67]。(2)精确编辑效率低。(3)编辑产物异质性高。DNA双链断裂经非同源末端连接修复时可产生40种[35],甚至更多的基因型,导致基因编辑产物异质性高,难以进行质控。
针对这些问题,可从以下几个方面进行研究和解决。
(1)改造递送系统提高编辑效率,降低脱靶率。质粒、病毒、mRNA和核糖核蛋白复合体(ribonucleoprotein, RNP)是依次开发出的四种CRISPR/Cas9递送系统[68](图1)。最新的核糖核蛋白复合体由向导RNA与Cas9蛋白体外孵育组装而成,进入细胞后发挥短时的基因编辑功能,有较高的基因编辑效率和更低的细胞毒性和脱靶率,更适用于基因治疗,但其转导效率有待进一步提升[45,68,69]。上述3项临床研究中(NCT02793856、NCT03399448、NCT03164135)有两项(NCT03399448、NCT03164135)使用核糖核蛋白复合体递送系统。
(2)改造CRISPR/Cas9系统提高编辑效率,降低脱靶率。在改造向导RNA方面,对于同一靶点不同向导RNA介导的基因编辑效率和脱靶率差异显著[70],目前主要通过实验筛选的方法,筛出效率最高、脱靶率最低的向导RNA进行后续实验。针对人和小鼠基因组,科学家基于大量实验开发了向导RNA库,用于评估向导RNA的编辑效果[70];对于mRNA和核糖核蛋白复合体递送系统,向导RNA的稳定性对基因编辑效率影响较大,化学修饰向导RNA可提升向导RNA稳定性,防止其过快降解,提升基因编辑效率,将插入或缺失(indel)率从2.4%提升至83.3%,同源重组效率从~15%提升至50%[71]。在改造Cas9蛋白方面,改造Cas9蛋白的氨基酸组成可提高靶向编辑率:总编辑率的比值,如p.R691A单点突变的HiFi Cas9,其总编辑效率与野生型Cas9相近,但on-target效率可达总编辑效率的99%,显著高于野生型的28%~72%,且HiFi Cas9较野生型Cas9脱靶率降低近20倍[72];在Cas9蛋白上融合核定位序列,辅助Cas9蛋白入核,也可提升基因编辑效率[45]。
(3)阐明DNA损伤修复机制。CRISPR/Cas9介导的基因编辑依赖DNA损伤修复机制,目前主要考虑同源重组和非同源末端连接。研究表明,微同源末端连接(microhomology-mediated end joining, MMEJ)是DNA损伤修复的又一重要途径,为基因编辑介导的基因治疗策略提供了更多可能[73]。
(4)找寻合适的基因编辑靶点。基因编辑安全实施的关键是找到特异且易编辑的靶点,而找寻靶点的基础是阐明疾病致病机制,使得基因治疗靶点不限于突变位点,而是可以拓展到突变基因的上下游或替代/补偿途径去寻找最合适的编辑靶点,比如,对于β-地贫和镰状细胞贫血,可通过重激活内源γ-珠蛋白基因,缓解疾病症状。该方法不受突变位点影响,适用范围广,为其他有类似调节机制的疾病提供了基因治疗的新思路。基因调控元件作为编辑靶点是近两年的研究趋势,越来越多的研究表明对启动子、增强子等进行编辑可有效调控基因表达,尤其对于谱系特异的调控元件,安全性更高。
(5)发展造血干细胞相关技术。血液病的基因治疗离不开干细胞技术的发展。目前血液病治疗和临床研究主要采用造血干细胞和祖细胞及其分化出的T淋巴细胞。造血干细胞和祖细胞的应用仍存在难以体外增殖、体外培养体系不成熟、已有分子标记特异性不强、同源重组修复效率低且不稳定等问题。基因编辑细胞移植至免疫缺陷小鼠后编辑效率降低可能就由于其中混有未编辑的造血干细胞和祖细胞[45],而目前基因编辑效率难以实现100%,也难以分选出正确编辑的造血干细胞和祖细胞。但造血干细胞和祖细胞安全性好,移植技术成熟,是基因治疗靶细胞的首选。
(6)发展诱导多能干细胞相关技术。2006年诱导多能干细胞(iPSC)的问世为遗传病基因治疗研究提供了良好的细胞模型,患者来源的iPSC与患者遗传背景一致,无伦理问题,且可在体外大量扩增。研究发现,CRISPR/Cas9在na ̈ıve iPSC中的编辑效率高出primed iPSC近两倍[74];在iPSC培养物中添加小分子化合物,同源重组修复效率可从6%提升至54%,同时抑制非同源末端连接的发生,这些研究可为造血干细胞和祖细胞基因编辑提供指导[30]。iPSC具有分化为三胚层细胞的潜能,但未分化的iPSC有致瘤风险,无法直接用于基因治疗[75]。将其分化为造血干细胞等成体干细胞或可解决此问题。目前将iPSC分化为造血干细胞和祖细胞的分化体系尚不成熟,分化出的造血干细胞在免疫缺陷小鼠中难以长期植入[76],基因组不稳定,易发生白血病转化[77]。此外,虽然实验室中突变修复后的β-地贫患者来源iPSC红系分化效率提高且分化所得红系细胞β-珠蛋白表达提升[15,28,29],但多数研究中突变未修复iPSC和突变修复iPSC来源的红系细胞均主要表达γ-珠蛋白[15,29,78],难以模拟正常人红系细胞主要表达β-珠蛋白的真实情况。
(7)其他。目前CRISPR/Cas9用于基因治疗的技术标准、临床疗效、不良反应等尚不明确,需待进一步的临床研究。基因编辑技术用于基因治疗的相关法律法规也有待进一步完善。
综上所述,基于CRISPR/Cas9技术的基因治疗研究主要集中于血液系统疾病,包括血液肿瘤和遗传性血液病,尤其适用于主要由单基因点突变或移码突变造成的β-地贫等疾病,最安全有效的细胞是患者自体造血干细胞和祖细胞,更安全的途径是体外途径。在体外造血干细胞和祖细胞处于G0期,主要采取非同源末端连接的方式修复DNA双链断裂,因此CRISPR/Cas9虽可修复多种类型突变,但更适用于内含子突变的删除和基因表达调控,尤其利用CRISPR/Cas9重激活γ-珠蛋白基因表达的基因治疗方式,已开始临床研究。但通过非同源末端连接的方式实现基因编辑终究无法精确修复基因突变,潜在的风险无法评估,因此最理想最希望实现的仍是通过同源重组修复精确修复基因突变,提高同源重组修复效率应是今后CRISPR/Cas9用于疾病治疗的重点研究方向。此外,单碱基编辑技术能实现高效且精确的单碱基编辑,在单碱基突变引起的疾病中有巨大的应用前景,可与CRISPR/Cas9技术互为补充。目前单碱基编辑技术进入临床要解决的首要问题亦是脱靶率高。相信随着上述问题的解决,基于CRISPR技术的基因治疗会成为安全且高效的基因治疗方法,为治愈遗传性疾病带来希望。
[1] Taher AT, Weatherall DJ, Cappellini MD. Thalassaemia.,2018, 391(10116): 155–167.
[2] Farashi S, Harteveld CL. Molecular basis of α-thalassemia.,2018, 70: 43–53.
[3] Zeng YT, Huang SZ. Disorders of haemoglobin in China.,1987, 24(10): 578–583.
[4] Liu JW, Hong T, Qin X, Liang YM, Zhang P. Recent advance on genome editing for therapy of β-hemoglobinopathies.,2018, 40(2): 95–103.刘佳伟, 洪涛, 秦鑫, 梁英民, 张萍. β-血红蛋白病基因组编辑治疗的研究进展. 遗传, 2018, 40(2): 95–103.
[5] Santagostino E, Fasulo MR. Hemophilia A and hemophilia B: different types of diseases?,2013, 39(7): 697–701.
[6] Blaese RM, Culver KW, Miller AD, Carter CS, Fleisher T, Clerici M, Shearer G, Chang L, Chiang Y, Tolstoshev P, Greenblatt JJ, Rosenberg SA, Klein H, Berger M, Mullen CA, Ramsey WJ, Muul L, Morgan RA, Anderson WF. T lymphocyte-directed gene therapy for ADA- SCID: initial trial results after 4 years.,1995, 270(5235): 475–480.
[7] Niu XR, Yin SM, Chen X, Shao TT, Li DL. Gene editing technology and its recent progress in disease therapy.,2019, 41(7): 582–598.牛煦然, 尹树明, 陈曦, 邵婷婷, 李大力. 基因编辑技术及其在疾病治疗中的研究进展. 遗传, 2019, 41(7): 582–598.
[8] Nathwani AC, Davidoff AM, Tuddenham EGD. Advances in gene therapy for hemophilia.,2017, 28(11): 1004–1012.
[9] Peyvandi F, Garagiola I. Clinical advances in gene therapy updates on clinical trials of gene therapy in haemophilia.,2019, 25(5): 738–746.
[10] Zeng YT, Huang SZ, Ren ZR, Lu ZH, Zeng FY, Schechter AN, Rodgers GP. Hydroxyurea therapy in beta-thalassaemia intermedia: improvement in haematological parameters due to enhanced beta-globin synthesis.,1995, 90(3): 557–563.
[11] Xie SY, Li W, Ren ZR, Huang SZ, Zeng FY, Zeng YT. Correction of β654-thalassaemia mice using direct intravenous injection of siRNA and antisense RNA vectors.,2011, 93(3): 301–310.
[12] Xie SY, Ren ZR, Zhang JZ, Guo XB, Wang QX, Wang S, Lin D, Gong XL, Li W, Huang SZ, Zeng FY, Zeng YT. Restoration of the balanced alpha/beta-globin gene expression in beta654-thalassemia mice using combined RNAi and antisense RNA approach.,2007, 16(21): 2616–2625.
[13] Davis R, Gurumurthy A, Hossain MA, Gunn EM, Bungert J. Engineering globin gene expression.,2018, 12: 102–110.
[14] Elalfy MS, Adly AAM, Ismail EA, Elhenawy YI, Elghamry IR. Therapeutic superiority and safety of combined hydroxyurea with recombinant human erythropoietin over hydroxyurea in young β-thalassemia intermedia patients.,2013, 91(6): 522–533.
[15] Xie F, Ye L, Chang JC, Beyer AI, Wang JM, Muench MO, Kan YW. Seamless gene correction of β-thalassemia mutations in patient-specific iPSCs using CRISPR/Cas9 and.,2014, 24(9): 1526–1533.
[16] Ren YX, Xiao RD, Lou XM, Fang XD. Research advance and application in the gene therapy of gene editing technologies.,2019, 41(1): 18–27.任云晓, 肖茹丹, 娄晓敏, 方向东. 基因编辑技术及其在基因治疗中的应用. 遗传, 2019, 41(1): 18–27.
[17] Garneau JE, Dupuis MÈ, Villion M, Romero DA, Barrangou R, Boyaval P, Fremaux C, Horvath P, Magadán AH, Moineau S. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA.,2010, 468(7320): 67–71.
[18] Deltcheva E, Chylinski K, Sharma CM, Gonzales K, Chao YJ, Pirzada ZA, Eckert MR, Vogel J, Charpentier E. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III.,2011, 471(7340): 602–607.
[19] Jinek M, Chylinski K, Fonfara I, Haue M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity.,2012, 337(6096): 816–821.
[20] Mali P, Yang LH, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. RNA-guided human genome engineeringCas9.,2013, 339(6121): 823–826.
[21] Zhang JH, Adikaram P, Pandey M, Genis A, Simonds WF. Optimization of genome editing through CRISPR-Cas9 engineering.,2016, 7(3): 166–174.
[22] Chang HHY, Pannunzio NR, Adachi N, Lieber MR. Non-homologous DNA end joining and alternative pathways to double-strand break repair.,2017, 18(8): 495–506.
[23] Li GL, Zhong CL, Mo JX, Quan R, Wu ZF, Li ZC, Yang HQ, Zhang XW. Advances in site-specific integration of transgene in animal genome.,2017, 39(2): 98–109.李国玲, 钟翠丽, 莫健新, 全绒, 吴珍芳, 李紫聪, 杨化强, 张献伟. 动物基因组定点整合转基因技术研究进展. 遗传, 2017, 39(2): 98–109.
[24] Zong Y, Gao CX. Progress on base editing systems.,2019, 41(9): 777–800.宗媛, 高彩霞. 碱基编辑系统研究进展. 遗传, 2019, 41(9): 777–800.
[25] Liang PP, Sun HW, Sun Y, Zhang XY, Xie XW, Zhang JR, Zhang Z, Chen YX, Ding CH, Xiong YY, Ma WB, Liu D, Huang JJ, Zhou SY. Effective gene editing by high-fidelity base editor 2 in mouse zygotes.,2017, 8(8): 601–611.
[26] Kim K, Ryu SM, Kim ST, Baek G, Kim D, Lim K, Chung E, Kim S, Kim JS. Highly efficient RNA-guided base editing in mouse embryos.,2017, 35(5): 435–437.
[27] Lai KT, Huang GF, Su L, He YY. The prevalence of thalassemia in mainland China: evidence from epidemiological surveys.,2017, 7(1): 920.
[28] Niu XH, He WY, Song B, Ou ZH, Fan D, Chen YC, Fan Y, Sun XF. Combining single strand oligodeoxynucleotides and CRISPR/Cas9 to correct gene mutations in β-thalassemia-induced pluripotent stem cells.,2016, 291(32): 16576–16585.
[29] Song B, Fan Y, He WY, Zhu DT, Niu XH, Wang D, Ou ZH, Luo M, Sun XF. Improved hematopoietic differentiation efficiency of gene-corrected beta-thalassemia induced pluripotent stem cells by CRISPR/Cas9 system.,2015, 24(9): 1053–1065.
[30] Liu YL, Yang Y, Kang XJ, Lin B, Yu Q, Song B, Gao G, Chen YY, Sun XF, Li XP, Bu L, Fan Y. One-step biallelic and scarless correction of a β-thalassemia mutation in patient-specific iPSCs without drug selection.,2017, 6: 57–67.
[31] Park SH, Lee CM, Dever DP, Davis TH, Camarena J, Srifa W, Zhang YK, Paikari A, Chang AK, Porteus MH, Sheehan VA, Bao G. Highly efficient editing of the β-globin gene in patient-derived hematopoietic stem and progenitor cells to treat sickle cell disease.,2019, 47(15): 7955–7972.
[32] Wattanapanitch M, Damkham N, Potirat P, Trakarnsanga K, Janan M, U-pratya Y, Kheolamai P, Klincumhom N, Issaragrisil S. One-step genetic correction of hemoglobin E/beta-thalassemia patient-derived iPSCs by the CRISPR/Cas9 system.,2018, 9(1): 46.
[33] Antony JS, Latifi N, Haque AKMA, Lamsfus-Calle A, Daniel-Moreno A, Graeter S, Baskaran P, Weinmann P, Mezger M, Handgretinger R, Kormann MSD. Gene correction of HBB mutations in CD34+hematopoietic stem cells using Cas9 mRNA and ssODN donors.,2018, 5(1): 9.
[34] Fang YD, Cheng Y, Lu D, Gong XL, Yang GH, Gong ZJ, Zhu YW, Sang X, Fan SY, Zhang JZ, Zeng FY. Treatment of β654-thalassaemia by TALENs in a mouse model.,2018, 51(6): e12491.
[35] Xu SQ, Luk K, Yao QM, Shen AH, Zeng J, Wu YX, Luo HY, Brendel C, Pinello L, Chui DHK, Wolfe SA, Bauer DE. Editing aberrant splice sites efficiently restores β-globin expression in β-thalassemia.,2019, 133(21): 2255–2262.
[36] Park CY, Kim DH, Son JS, Sung JJ, Lee J, Bae S, Kim JH, Kim DW, Kim JS. Functional correction of large factor VIII gene chromosomal inversions in hemophilia A patient-derived iPSCs using CRISPR-Cas9.,2015, 17(2): 213–220.
[37] Peng Z, Zhou WC, Fu WQ, Du RQ, Jin L, Zhang F. Correlation between frequency of non-allelic homologous recombination and homology properties: evidence from homology-mediated CNV mutations in the human genome.,2015, 24(5): 1225–1233.
[38] Sasaki M, Lange J, Keeney S. Genome destabilization by homologous recombination in the germ line.,2010, 11(3): 182–195.
[39] Park CY, Kim J, Kweon J, Son JS, Lee JS, Yoo JE, Cho SR, Kim JH, Kim JS, Kim DW. Targeted inversion and reversion of the blood coagulation factor 8 gene in human iPS cells using TALENs.,2014, 111(25): 9253–9258.
[40] George CM, Alani E. Multiple cellular mechanisms prevent chromosomal rearrangements involving repetitive DNA.,2012, 47(3): 297–313.
[41] Zeng J, Wu YX, Ren CY, Bonanno J, Shen AH, Shea D, Gehrke JM, Clement K, Luk K, Yao QM, Kim R, Wolfe SA, Manis JP, Pinello L, Joung JK, Bauer DE. Therapeutic base editing of human hematopoietic stem cells.,2020, 26(4): 535–541.
[42] Zhou CY, Sun YD, Yan R, Liu YJ, Zuo EW, Gu C, Han LX, Wei Y, Hu XD, Zeng R, Li YX, Zhou HB, Guo F, Yang H. Off-target RNA mutation induced by DNA base editing and its elimination by mutagenesis.,2019, 571(7764): 275–278.
[43] Thein SL. Molecular basis of β thalassemia and potential therapeutic targets.,2018, 70: 54–65.
[44] Mettananda S, Fisher CA, Hay D, Badat M, Quek L, Clark K, Hublitz P, Downes D, Kerry J, Gosden M, Telenius J, Sloane-Stanley JA, Faustino P, Coelho A, Doondeea J, Usukhbayar B, Sopp P, Sharpe JA, Hughes JR, Vyas P, Gibbons RJ, Higgs DR. Editing an α-globin enhancer in primary human hematopoietic stem cells as a treatment for β-thalassemia.,2017, 8(1): 424.
[45] Wu YX, Zeng J, Roscoe BP, Liu PP, Yao QM, Lazzarotto CR, Clement K, Cole MA, Luk K, Baricordi C, Shen AH, Ren CY, Esrick EB, Manis JP, Dorfman DM, Williams DA, Biffi A, Brugnara C, Biasco L, Brendel C, Pinello L, Tsai SQ, Wolfe SA, Bauer DE. Highly efficient therapeutic gene editing of human hematopoietic stem cells.,2019, 25(5): 776–783.
[46] Martyn GE, Wienert B, Yang L, Shah M, Norton LJ, Burdach J, Kurita R, Nakamura Y, Pearson RCM, Funnell APW, Quinlan KGR, Crossley M. Natural regulatory mutations elevate the fetal globin genedisruption of BCL11A or ZBTB7A binding.,2018, 50(4): 498–503.
[47] Martyn GE, Wienert B, Kurita R, Nakamura Y, Quinlan KGR, Crossley M. A natural regulatory mutation in the proximal promoter elevates fetal globin expression by creating a de novo GATA1 site.,2019, 133(8): 852–856.
[48] Antoniani C, Meneghini V, Lattanzi A, Felix T, Romano O, Magrin E, Weber L, Pavani G, Hoss SE, Kurita R, Nakamura Y, Cradick TJ, Lundberg AS, Porteus M, Amendola M, Nemer WE, Cavazzana M, Mavilio F, Miccio A. Induction of fetal hemoglobin synthesis by CRISPR/Cas9-mediated editing of the human β-globin locus.,2018, 131(17): 1960–1973.
[49] Canver MC, Smith EC, Sher F, Pinello L, Sanjana NE, Shalem O, Chen DD, Schupp PG, Vinjamur DS, Garcia SP, Luc S, Kurita R, Nakamura Y, Fujiwara Y, Maeda T, Yuan GC, Zhang F, Orkin SH, Bauer DE. BCL11A enhancer dissection by Cas9-mediatedsaturating mutagenesis.,2015, 527(7577): 192–197.
[50] Liu PT, Keller JR, Ortiz M, Tessarollo L, Rachel RA, Nakamura T, Jenkins NA, Copeland NG. Bcl11a is essential for normal lymphoid development.,2003, 4(6): 525–532.
[51] Li J, Lai YR, Shi LL. BCL11A down-regulation induces γ-globin in human β-thalassemia major erythroid cells.,2018, 42(4): 225–230.
[52] Wang LR, Li LX, Ma YL, Hu HD, Li Q, Yang Y, Liu WB, Yin SM, Li W, Fu B, Kurita R, Nakamura Y, Liu M, Lai YR, Li DL. Reactivation of γ-globin expression through Cas9 or base editor to treat β-hemoglobinopathies.,2020, 30(3): 276–278.
[53] Grevet JD, Lan XJ, Hamagami N, Edwards CR, Sankaranarayanan L, Ji XJ, Bhardwaj SK, Face CJ, Posocco DF, Abdulmalik O, Keller CA, Giardine B, Sidoli S, Garcia BA, Chou ST, Liebhaber SA, Hardison RC, Shi JW, Blobel GA. Domain-focused CRISPR screen identifies HRI as a fetal hemoglobin regulator in human erythroid cells.,2018, 361(6399): 285–290.
[54] Thompson AA, Walters MC, Kwiatkowski J, Rasko JEJ, Ribeil JA, Hongeng S, Magrin E, Schiller GJ, Payen E, Semeraro M, Moshous D, Lefrere F, Puy H, Bourget P, Magnani A, Caccavelli L, Diana JS, Suarez F, Monpoux F, Brousse V, Poirot C, Brouzes C, Meritet JF, Pondarré C, Beuzard Y, Chrétien S, Lefebvre T, Teachey DT, Anurathapan U, Ho PJ, von Kalle C, Kletzel M, Vichinsky E, Soni S, Veres G, Negre O, Ross RW, Davidson D, Petrusich A, Sandler L, Asmal M, Hermine O, Montalembert MD, Hacein-Bey-Abina S, Blanche S, Leboulch P, Cavazzana M. Gene therapy in patients with transfusion-dependent β-thalassemia.,2018, 378(16): 1479–1493.
[55] Chen YH, Keiser MS, Davidson BL. Viral vectors for gene transfer.,2018, 8(4): e58.
[56] Cunningham SC, Dane AP, Spinoulas A, Alexander IE. Gene delivery to the juvenile mouse liver using AAV2/8 vectors.,2008, 16(6): 1081–1088.
[57] George LA, Sullivan SK, Giermasz A, Rasko JEJ, Samelson-Jones BJ, Ducore J, Cuker A, Sullivan LM, Majumdar S, Teitel J, McGuinn CE, Ragni MV, Luk AY, Hui D, Wright JF, Chen YF, Liu Y, Wachtel K, Winters A, Tiefenbacher S, Arruda VR, van der Loo JCM, Zelenaia O, Takefman D, Carr ME, Couto LB, Anguela XM, High KA. Hemophilia B gene therapy with a high-specific-activity factor IX variant.,2017, 377(23): 2215– 2227.
[58] Wang LL, Yang Y, Breton CA, White J, Zhang J, Che Y, Saveliev A, McMenamin D, He ZN, Latshaw C, Li MY, Wilson JM. CRISPR/Cas9-mediatedgene targeting corrects hemostasis in newborn and adult factor IX-knockout mice.,2019, 133(26): 2745–2752.
[59] Baliou S, Adamaki M, Kyriakopoulos AM, Spandidos DA, Panayiotidis M, Christodoulou I, Zoumpourlis V. CRISPR therapeutic tools for complex genetic disorders and cancer (Review).,2018, 53(2): 443–468.
[60] Cyranoski D. CRISPR gene-editing tested in a person for the first time.,2016, 539(7630): 479.
[61] Lu Y, Xue JX, Deng T, Zhou XJ, Yu K, Deng L, Huang MJ, Yi X, Liang MZ, Wang Y, Shen HG, Tong RZ, Wang WB, Li L, Song J, Li J, Su XX, Ding ZY, Gong YL, Zhu J, Wang YS, Zou BW, Zhang Y, Li YY, Zhou L, Liu YM, Yu M, Wang YQ, Zhang XW, Yin LM, Xia XF, Zeng Y, Zhou Q, Ying BW, Chen C, Wei YQ, Li WM, Mok T. Safety and feasibility of CRISPR-edited T cells in patients with refractory non-small-cell lung cancer.,2020, 26(5): 732–740.
[62] Stadtmauer EA, Fraietta JA, Davis MM, Cohen AD, Weber KL, Lancaster E, Mangan PA, Kulikovskaya I, Gupta M, Chen F, Tian LF, Gonzalez VE, Xu J, Jung IY, Melenhorst JJ, Plesa G, Shea J, Matlawski T, Cervini A, Gaymon AL, Desjardins S, Lamontagne A, Salas-Mckee J, Fesnak A, Siegel DL, Levine BL, Jadlowsky JK, Young RM, Chew A, Hwang WT, Hexner EO, Carreno BM, Nobles CL, Bushman FD, Parker KR, Qi YY, Satpathy AT, Chang HY, Zhao YB, Lacey SF, June CH. CRISPR-engineered T cells in patients with refractory cancer.,2020, 367(6481): eaba7365.
[63] Xu L, Wang J, Liu YL, Xie LF, Su B, Mou DL, Wang LT, Liu TT, Wang XB, Zhang B, Zhao L, Hu LD, Ning HM, Zhang YF, Deng K, Liu LF, Lu XF, Zhang T, Xu J, Li C, Wu H, Deng HK, Chen H. CRISPR-edited stem cells in a patient with HIV and acute lymphocytic leukemia.,2019, 381(13): 1240–1247.
[64] Wang DW, Wang K, Cai YJ. An overview of development in gene therapeutics in China.,2020, 27(7–8): 338–348.
[65] Park JH, Geyer MB, Brentjens RJ. CD19-targeted CAR T-cell therapeutics for hematologic malignancies: interpreting clinical outcomes to date.,2016, 127(26): 3312– 3320.
[66] Fu YF, Foden JA, Khayter C, Maeder ML, Reyon D, Joung JK, Sander JD. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells.,2013, 31(9): 822–826.
[67] Cullot G, Boutin J, Toutain J, Prat F, Pennamen P, Rooryck C, Teichmann M, Rousseau E, Lamrissi-Garcia I, Guyonnet-Duperat V, Bibeyran A, Lalanne M, Prouzet- Mauléon V, Turcq B, Ged C, Blouin JM, Richard E, Dabernat S, Moreau-Gaudry F, Bedel A. CRISPR-Cas9 genome editing induces megabase-scale chromosomal truncations.,2019, 10(1): 1136.
[68] Lattanzi A, Meneghini V, Pavani G, Amor F, Ramadier S, Felix T, Antoniani C, Masson C, Alibeu O, Lee C, Porteus MH, Bao G, Amendola M, Mavilio F, Miccio A. Optimization of CRISPR/Cas9 delivery to human hematopoietic stem and progenitor cells for therapeutic genomic rearrangements.,2019, 27(1): 137–150.
[69] Dever DP, Bak RO, Reinisch A, Camarena J, Washington G, Nicolas CE, Pavel-Dinu M, Saxena N, Wilkens AB, Mantri S, Uchida N, Hendel A, Narla A, Majeti R, Weinberg KI, Porteus MH. CRISPR/Cas9 β-globin gene targeting in human haematopoietic stem cells.,2016, 539(7629): 384–389.
[70] Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, Smith I, Tothova Z, Wilen C, Orchard R, Virgin HW, Listgarten J, Root DE. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9.,2016, 34(2): 184–191.
[71] Hendel A, Bak RO, Clark JT, Kennedy AB, Ryan DE, Roy S, Steinfeld I, Lunstad BD, Kaiser RJ, Wilkens AB, Bacchetta R, Tsalenko A, Dellinger D, Bruhn L, Porteus MH. Chemically modified guide RNAs enhance CRISPR- Cas genome editing in human primary cells.,2015, 33(9): 985–989.
[72] Vakulskas CA, Dever DP, Rettig GR, Turk R, Jacobi AM, Collingwood MA, Bode NM, McNeill MS, Yan SQ, Camarena J, Lee CM, Park SH, Wiebking V, Bak RO, Gomez-Ospina N, Pavel-Dinu M, Sun WC, Bao G, Porteus MH, Behlke MA. A high-fidelity Cas9 mutant delivered as a ribonucleoprotein complex enables efficient gene editing in human hematopoietic stem and progenitor cells.,2018, 24(8): 1216–1224.
[73] Shen MW, Arbab M, Hsu JY, Worstell D, Culbertson SJ, Krabbe O, Cassa CA, Liu DR, Gifford DK, Sherwood RI. Predictable and precise template-free CRISPR editing of pathogenic variants.,2018, 563(7733): 646–651.
[74] Yang YY, Zhang XB, Yi L, Hou ZZ, Chen JY, Kou XC, Zhao YH, Wang H, Sun XF, Jiang CZ, Wang YX, Gao SR. Naïve induced pluripotent stem cells generated from β-thalassemia fibroblasts allow efficient gene correction with CRISPR/Cas9.,2016, 5(2): 267.
[75] Volarevic V, Markovic BS, Gazdic M, Volarevic A, Jovicic N, Arsenijevic N, Armstrong L, Djonov V, Lako M, Stojkovic M. Ethical and safety issues of stem cell-based therapy.,2018, 15(1): 36–45.
[76] Paes BCMF, Moço PD, Pereira CG, Porto GS, de Sousa Russo EM, Reis LCJ, Covas DT, Picanço-Castro V. Ten years of iPSC: clinical potential and advanceshematopoietic differentiation.,2017, 33(3): 233–250.
[77] Tan YT, Ye L, Xie F, Beyer AI, Muench MO, Wang JM, Chen Z, Liu H, Chen SJ, Kan YW. Respecifying human iPSC-derived blood cells into highly engraftable hematopoietic stem and progenitor cells with a single factor.,2018, 115(9): 2180–2185.
[78] Uchida N, Haro-Mora JJ, Fujita A, Lee DY, Winkler T, Hsieh MM, Tisdale JF. Efficient generation of β-globin- expressing erythroid cells using stromal cell-derived induced pluripotent stem cells from patients with sickle cell disease.,2017, 35(3): 586–596.
Advances in gene therapy for β-thalassemia and hemophilia based on the CRISPR/Cas9 technology
Liwen Bao1, Yiye Zhou2, Fanyi Zeng1,2
Thalassemia and hemophilia are common inherited blood disorders caused by genetic abnormalities. These diseases are difficult to cure and can be inherited to the next generation, causing severe family and social burden. The emergence of gene therapy provides a new treatment for genetic diseases. However, since its first clinical trial in 1990, the development of gene therapy has not been as optimistic in the past three decades as one could hope. The development of gene-editing technology, particularly the third generation gene-editing technology CRISPR/Cas9 (clustered regularly interspaced short palindromic repeats/CRISPR-associated protein 9), has given hope in such therapeutic approach for having advantages in high editing efficiency, simple operation, and low cost. Gene editing-mediated gene therapy has thus received increasing attention from the biomedical community. It has shown promises for the treatment of inherited blood disorders, such as thalassemia and hemophilia. This paper reviews the fundamental research progress of gene therapy for β-thalassemia and hemophilia based on CRISPR/Cas9 technology in the past six years. It also summarizes the CRISPR/Cas9-based clinical trials of gene therapy. The problems and possible solutions to this technology for gene therapy are also discussed, thereby providing a reference for the research on gene therapy of inherited blood disorders based on CRISPR/Cas9 technology.
CRISPR/Cas9; gene therapy; inherited blood disorders; clinical trials
2020-04-21;
2020-08-25
国家重点研发计划(编号:2016YFC1000503),上海市重中之重重点学科项目(编号:2017ZZ02019),上海市临床重点专科项目(编号:shslczdzk05705) [Supported by the National Key Research and Development Program of China (No. 2016YFC1000503), Shanghai Key Disciplines Program (No. 2017ZZ02019), Key Clinical Specialty Projects in Shanghai (No. shslczdzk05705)]
鲍莉雯,硕士研究生,专业方向:生物学。E-mail: blwbaoliwen@163.com
曾凡一,博士,研究员,研究方向:遗传学。E-mail: fzeng@vip.163.com
10.16288/j.yczz.20-110
2020/9/16 16:00:01
URI: https://kns.cnki.net/kcms/detail/11.1913.R.20200915.1721.002.html
(责任编委: 徐湘民)
猜你喜欢
检验医学与临床(2021年14期)2021-07-29 07:40:24
教学考试(高考生物)(2020年6期)2020-11-23 05:25:56
食品与生物技术学报(2020年8期)2020-01-06 08:00:56
科学24小时(2019年5期)2019-06-11 08:39:38
发明与创新(2019年9期)2019-03-26 02:22:48
华人时刊(2017年21期)2018-01-31 02:24:04
中国比较医学杂志(2018年5期)2018-01-22 13:21:04
海南医学(2016年8期)2016-06-08 05:43:00
检验医学与临床(2015年14期)2015-03-16 01:46:53
结直肠肛门外科(2015年6期)2015-02-26 03:12:49
